
Gut Mycobiome Changes During COVID-19 Disease

 , , , , , , , and add
Show full author list
, , , , , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. ITS Sequencing
2.2.1. DNA Isolation
2.2.2. NGS Library Preparation
2.3. Bioinformatic Analysis
3. Results
3.1. ASV Calling Results and Decontamination
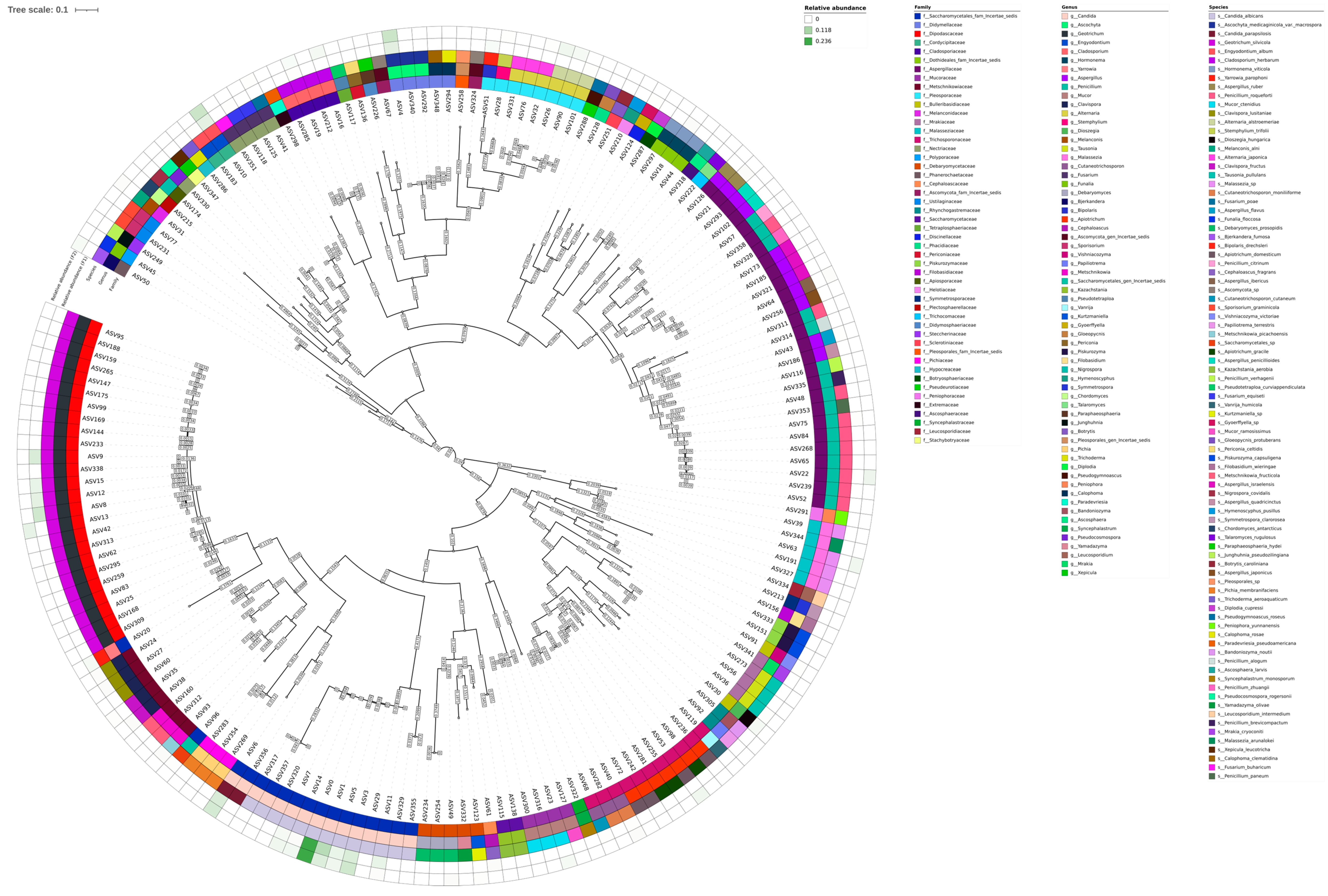
3.2. Taxonomic Composition Description
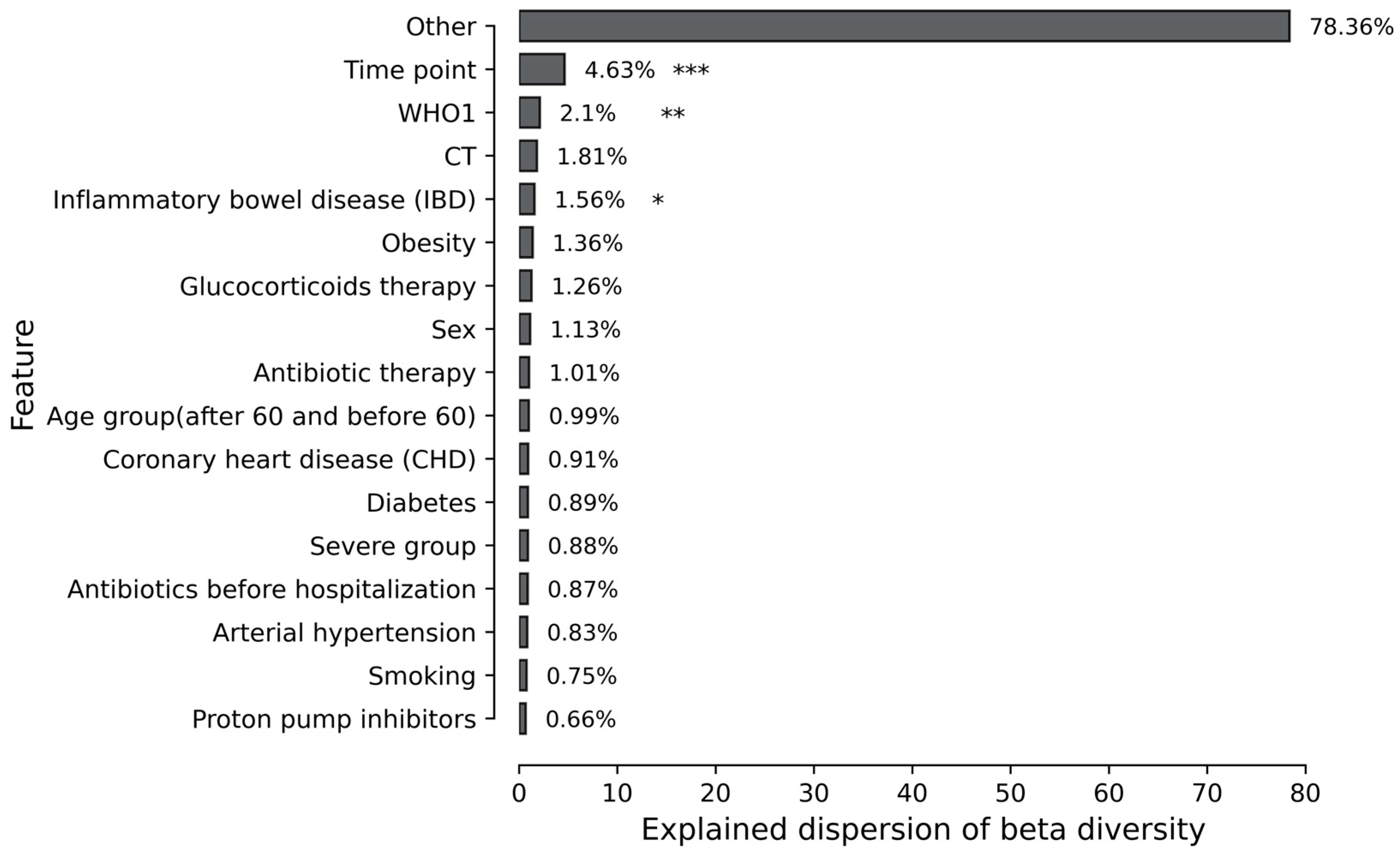
3.3. Association of Mycobiome with Patient Comorbidity and Treatment Procedure
3.4. Mycobiome Differences Across Time Points
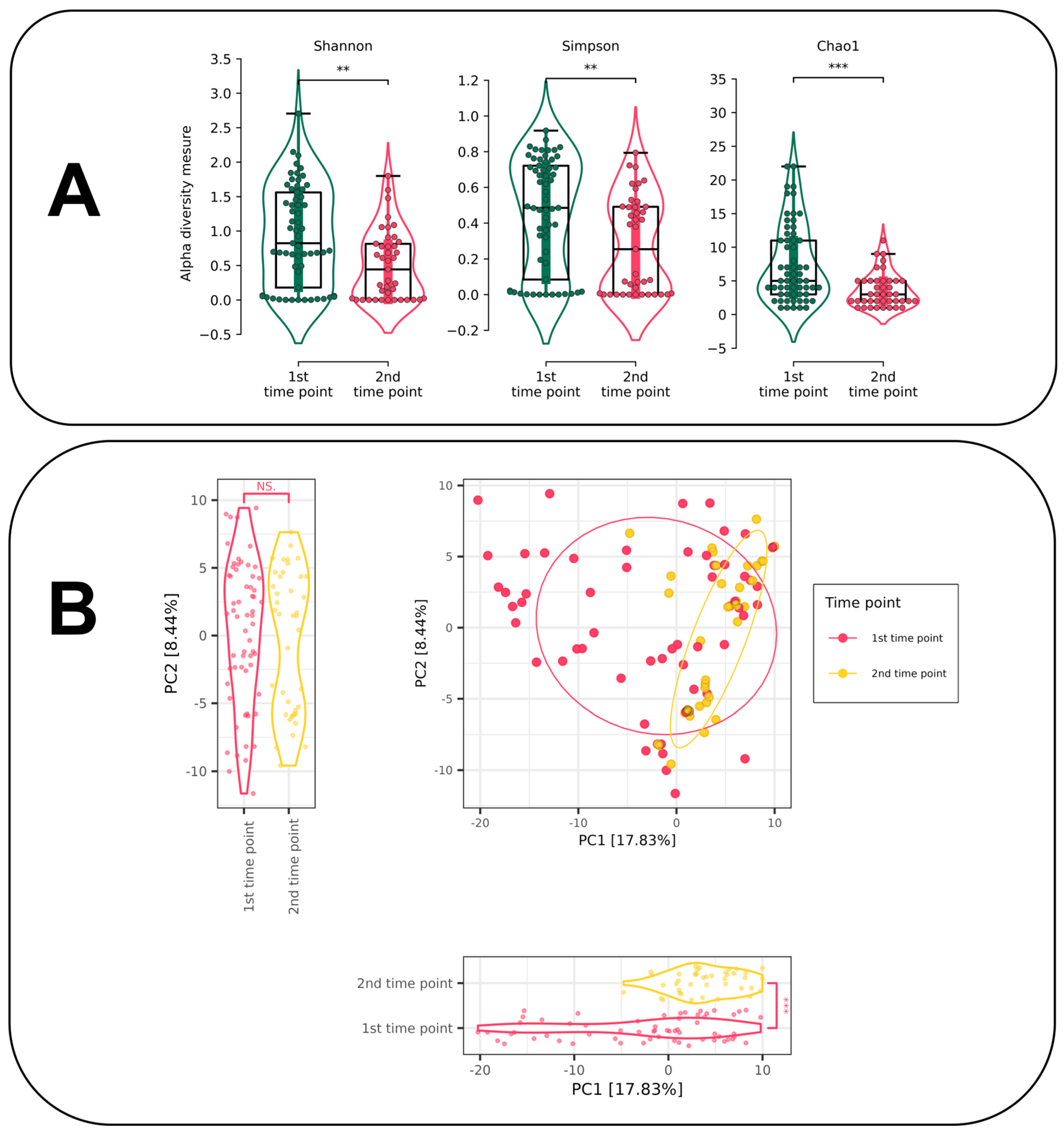
3.4.1. Alpha and Beta Diversity Estimation for Different Time Points
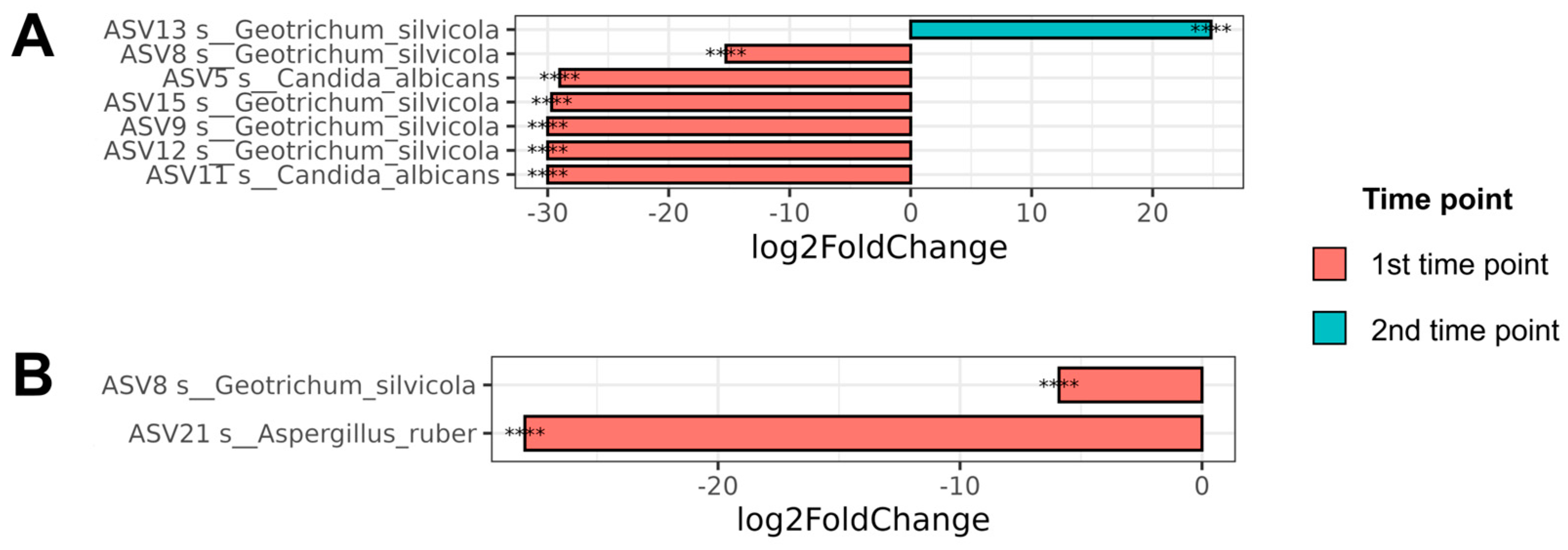
3.4.2. Differential Abundance Analysis for Different Time Points
3.5. Severity as a Factor of Fungal Alpha Diversity
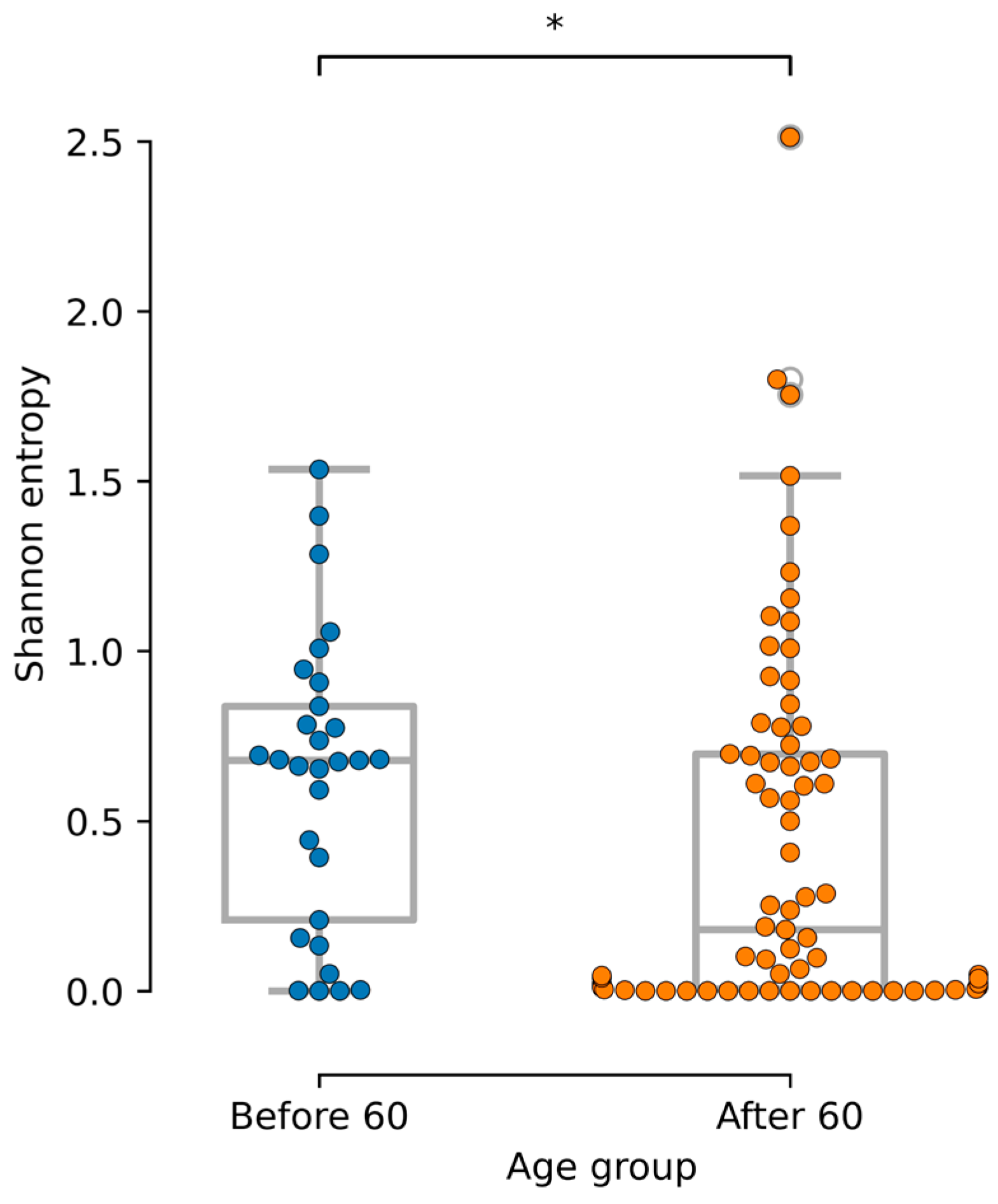
3.6. Age-Related Mycobiome Associations
3.7. Functional Annotation via FunFun
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koulenti, D.; Karvouniaris, M.; Paramythiotou, E.; Koliakos, N.; Markou, N.; Paranos, P.; Meletiadis, J.; Blot, S. Severe Candida infections in critically ill patients with COVID-19. J. Intensive Med. 2023, 3, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Hoenigl, M.; Seidel, D.; Sprute, R.; Cunha, C.; Oliverio, M.; Goldman, G.H.; Ibrahim, A.S.; Carvalho, A. COVID-19-associated fungal infections. Nat. Microbiol. 2022, 7, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhan, H.; Xu, W.; Yan, S.; Ng, S.C. The role of gut mycobiome in health and diseases. Ther. Adv. Gastroenterol. 2021, 14, 17562848211047130. [Google Scholar] [CrossRef]
- Zhang, F.; Aschenbrenner, D.; Yoo, J.Y.; Zuo, T. The gut mycobiome in health, disease, and clinical applications in association with the gut bacterial microbiome assembly. Lancet Microbe 2022, 3, e969–e983. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Yan, Y.; Pu, Y.; Lin, S.; Qiu, J.G.; Jiang, B.H.; Keller, M.I.; Wang, M.; Bork, P.; Chen, W.H.; et al. Enterotypes of the human gut mycobiome. Microbiome 2023, 11, 179. [Google Scholar] [CrossRef]
- Zuo, T.; Zhan, H.; Zhang, F.; Liu, Q.; Tso, E.Y.; Lui, G.C.Y.; Chen, N.; Li, A.; Lu, W.; Chan, F.K.L.; et al. Alterations in fecal fungal microbiome of patients with COVID-19 during time of hospitalization until discharge. Gastroenterology 2020, 159, 1302–1310. [Google Scholar] [CrossRef]
- Pal, R.; Singh, B.; Bhadada, S.K.; Banerjee, M.; Bhogal, R.S.; Hage, N.; Kumar, A. COVID-19-associated mucormycosis: An updated systematic review of literature. Mycoses 2021, 64, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Meijer, E.F.; Dofferhoff, A.S.; Hoiting, O.; Meis, J.F. COVID-19-associated pulmonary aspergillosis: A prospective single-center dual case series. Mycoses 2021, 64, 457–464. [Google Scholar] [CrossRef]
- Ren, Z.; Liu, S.; Wang, Q.; Rao, B.; Zeng, Z.; Xu, Y.; Wang, H.; Luo, H.; Gou, J.; Yu, Z. Alterations of the oral and gut mycobiome and cytokines during long-term follow-up of COVID-19 convalescents. Signal Transduct. Targeted Ther. 2023, 8, 166. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.D.; Lee, S.B.; Taylor, J.W.; Innis, M.A.; Gelfand, D.; Sninsky, J. Amplification and direct sequencing of fungal ribosomal RNA Genes for phylogenetics. In PCR Protocols; Academic Press, Inc.: Cambridge, MA, USA, 1990; pp. 315–322. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Rolling, T.; Zhai, B.; Frame, J.; Hohl, T.M.; Taur, Y. Customization of a DADA2-based pipeline for fungal internal transcribed spacer 1 (ITS1) amplicon data sets. JCI Insight 2022, 7, e151663. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; May, T.W.; Frøslev, T.G.; Pawlowska, J.; Lindahl, B.; Põldmaa, K.; Truong, C.; et al. The UNITE database for molecular identification and taxonomic communication of fungi and other eukaryotes: Sequences, taxa and classifications reconsidered. Nucleic Acids Res. 2024, 52, D791–D797. [Google Scholar] [CrossRef]
- Frøslev, T.G.; Kjøller, R.; Bruun, H.H.; Ejrnæs, R.; Brunbjerg, A.K.; Pietroni, C.; Hansen, A.J. Algorithm for post-clustering curation of DNA amplicon data yields reliable biodiversity estimates. Nat. Commun. 2017, 8, 1188. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Holmes, I.; Harris, K.; Quince, C. Dirichlet multinomial mixtures: Generative models for microbial metagenomics. PLoS ONE 2012, 7, e30126. [Google Scholar] [CrossRef]
- Krivonos, D.V.; Konanov, D.N.; Ilina, E.N. FunFun: ITS-based functional annotator of fungal communities. Ecol. Evol. 2023, 13, e9874. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Morozov, S.P.; Andreychenko, A.E.; Blokhin, I.A.; Gelezhe, P.B.; Gonchar, A.P.; Nikolaev, A.E.; Pavlov, N.A.; Chernina, V.Y.; Gombolevskiy, V.A. MosMedData: Data set of 1110 chest CT scans performed during the COVID-19 epidemic. Digit. Diagn. 2020, 1, 49–59. [Google Scholar] [CrossRef]
- Frappaz, D.; Bonneville-Levard, A.; Ricard, D.; Carrie, S.; Schiffler, C.; Xuan, K.H.; Weller, M. Assessment of Karnofsky (KPS) and WHO (WHO-PS) performance scores in brain tumour patients: The role of clinician bias. Support. Care Cancer 2021, 29, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Galeeva, J.S.; Fedorov, D.E.; Starikova, E.V.; Manolov, A.I.; Pavlenko, A.V.; Selezneva, O.V.; Klimina, K.M.; Veselovsky, V.A.; Morozov, M.D.; Yanushevich, O.O.; et al. Microbial Signatures in COVID-19: Distinguishing Mild and Severe Disease via Gut Microbiota. Biomedicines 2024, 12, 996. [Google Scholar] [CrossRef]
- Sam, Q.H.; Chang, M.W.; Chai, L.Y. The Fungal Mycobiome and Its Interaction with Gut Bacteria in the Host. Int. J. Mol. Sci. 2017, 18, 330. [Google Scholar] [CrossRef] [PubMed]
- Hamad, I.; Sokhna, C.; Raoult, D.; Bittar, F. Molecular detection of eukaryotes in a single human stool sample from Senegal. PLoS ONE 2012, 7, e40888. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, R.S.; Alves, P.D.; Corrêa, A.; Lachance, M.A.; Prasad, G.S.; Rajaram; Sinha, B.R.R.P.; Rosa, C.A. Geotrichum silvicola sp. nov., a novel asexual arthroconidial yeast species related to the genus Galactomyces. Int. J. Syst. Evol. Microbiol. 2005, 55, 497–501. [Google Scholar] [CrossRef]
- Zhu, H.Y.; Shang, Y.J.; Wei, X.Y.; Groenewald, M.; Robert, V.; Zhang, R.P.; Li, A.-H.; Han, P.-J.; Ji, F.; Li, J.-N.; et al. Taxonomic revision of Geotrichum and Magnusiomyces, with the descriptions of five new Geotrichum species from China. Mycology 2024, 15, 400–423. [Google Scholar] [CrossRef] [PubMed]
- Belvoncikova, P.; Splichalova, P.; Videnska, P.; Gardlik, R. The Human Mycobiome: Colonization, Composition and the Role in Health and Disease. J. Fungi 2022, 8, 1046. [Google Scholar] [CrossRef]
- Balasingham, S.; Chalkias, S.; Balasingham, A.; Saul, Z.; Wickes, B.L.; Sutton, D.A. A case of bovine valve endocarditis caused by Engyodontium album. Med. Mycol. 2011, 49, 430–434. [Google Scholar] [CrossRef]
- Thamke, D.C.; Mendiratta, D.K.; Dhabarde, A.; Shukla, A.K. Mycotic keratitis due to Engyodontium album: First case report from India. Indian J. Med. Microbiol. 2015, 33, 303–304. [Google Scholar] [CrossRef]
- Hallen-Adams, H.E.; Suhr, M.J. Fungi in the healthy human gastrointestinal tract. Virulence 2017, 8, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Zhou, W.; Duan, T.; Li, Y.; Matthew, C.; Nan, Z. Alfalfa Spring Black Stem and Leaf Spot Disease Caused by Phoma medicaginis: Epidemic Occurrence and Impacts. Microorganisms 2024, 12, 1279. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Jiang, J.R.; Zhang, G.Z.; Cai, L.; Crous, P.W. Resolving the Phoma enigma. Stud. Mycol. 2015, 82, 137–217. [Google Scholar] [CrossRef]
- Zhou, Y.; Cheng, L.; Lei, Y.L.; Ren, B.; Zhou, X. The Interactions Between Candida albicans and Mucosal Immunity. Front. Microbiol. 2021, 12, 652725. [Google Scholar] [CrossRef]
- Richardson, J.P.; Moyes, D.L. Adaptive immune responses to Candida albicans infection. Virulence 2015, 6, 327–337. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.; van der Meer, J.W.; Kullberg, B.J.; van de Veerdonk, F.L. Immune defence against Candida fungal infections. Nature reviews. Immunology 2015, 15, 630–642. [Google Scholar] [CrossRef]
- Román-Montes, C.M.; Sifuentes-Osornio, J.; Martínez-Gamboa, A. Cutaneous Infections by Geotrichum spp. Curr. Fungal Infect. Rep. 2024, 18, 60–68. [Google Scholar] [CrossRef]
- Pal, M.; Sejra, S.; Sejra, A.R.; Tesfaye, S. Geotrichosis—An Opportunistic Mycosis of Humans And Animals. Int. J. Livest. Res. 2013, 3, 38–44. [Google Scholar] [CrossRef]
- Lee, J.; Oh, S.J.; Ha, E.; Shin, G.Y.; Kim, H.J.; Kim, K.; Lee, C.K. Gut microbial and human genetic signatures of inflammatory bowel disease increase risk of comorbid mental disorders. NPJ Genom. Med. 2024, 9, 52. [Google Scholar] [CrossRef]
- Pisani, A.; Rausch, P.; Bang, C.; Ellul, S.; Tabone, T.; Marantidis Cordina, C.; Zahra, G.; Franke, A.; Ellul, P. Dysbiosis in the Gut Microbiota in Patients with Inflammatory Bowel Disease during Remission. Microbiol. Spectr. 2022, 10, e0061622. [Google Scholar] [CrossRef] [PubMed]
- Seelbinder, B.; Chen, J.; Brunke, S.; Vazquez-Uribe, R.; Santhaman, R.; Meyer, A.C.; de Oliveira Lino, F.S.; Chan, K.F.; Loos, D.; Imamovic, L.; et al. Antibiotics create a shift from mutualism to competition in human gut communities with a longer-lasting impact on fungi than bacteria. Microbiome 2020, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Lehallier, B.; Gate, D.; Schaum, N.; Nanasi, T.; Lee, S.E.; Yousef, H.; Moran Losada, P.; Berdnik, D.; Keller, A.; Verghese, J.; et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat. Med. 2019, 25, 1843–1850. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wang, C.; Zhou, X.; Zhou, W.; Hornburg, D.; Wu, S.; Snyder, M.P. Nonlinear dynamics of multi-omics profiles during human aging. Nat. Aging 2024, 4, 1619–1634. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krivonos, D.V.; Fedorov, D.E.; Klimina, K.M.; Veselovsky, V.A.; Kovalchuk, S.N.; Pavlenko, A.V.; Yanushevich, O.O.; Andreev, D.N.; Sokolov, F.S.; Fomenko, A.K.; et al. Gut Mycobiome Changes During COVID-19 Disease. J. Fungi 2025, 11, 194. https://doi.org/10.3390/jof11030194
Krivonos DV, Fedorov DE, Klimina KM, Veselovsky VA, Kovalchuk SN, Pavlenko AV, Yanushevich OO, Andreev DN, Sokolov FS, Fomenko AK, et al. Gut Mycobiome Changes During COVID-19 Disease. Journal of Fungi. 2025; 11(3):194. https://doi.org/10.3390/jof11030194
Chicago/Turabian StyleKrivonos, Danil V., Dmitry E. Fedorov, Ksenia M. Klimina, Vladimir A. Veselovsky, Svetlana N. Kovalchuk, Alexander V. Pavlenko, Oleg O. Yanushevich, Dmitry N. Andreev, Filipp S. Sokolov, Aleksey K. Fomenko, and et al. 2025. "Gut Mycobiome Changes During COVID-19 Disease" Journal of Fungi 11, no. 3: 194. https://doi.org/10.3390/jof11030194
APA StyleKrivonos, D. V., Fedorov, D. E., Klimina, K. M., Veselovsky, V. A., Kovalchuk, S. N., Pavlenko, A. V., Yanushevich, O. O., Andreev, D. N., Sokolov, F. S., Fomenko, A. K., Devkota, M. K., Andreev, N. G., Zaborovsky, A. V., Tsaregorodtsev, S. V., Evdokimov, V. V., Krikheli, N. I., Bely, P. A., Levchenko, O. V., Maev, I. V., ... Ilina, E. N. (2025). Gut Mycobiome Changes During COVID-19 Disease. Journal of Fungi, 11(3), 194. https://doi.org/10.3390/jof11030194