FungiDB: An Integrated Bioinformatic Resource for Fungi and Oomycetes

 , , ,
, , ,
Abstract
:1. Introduction
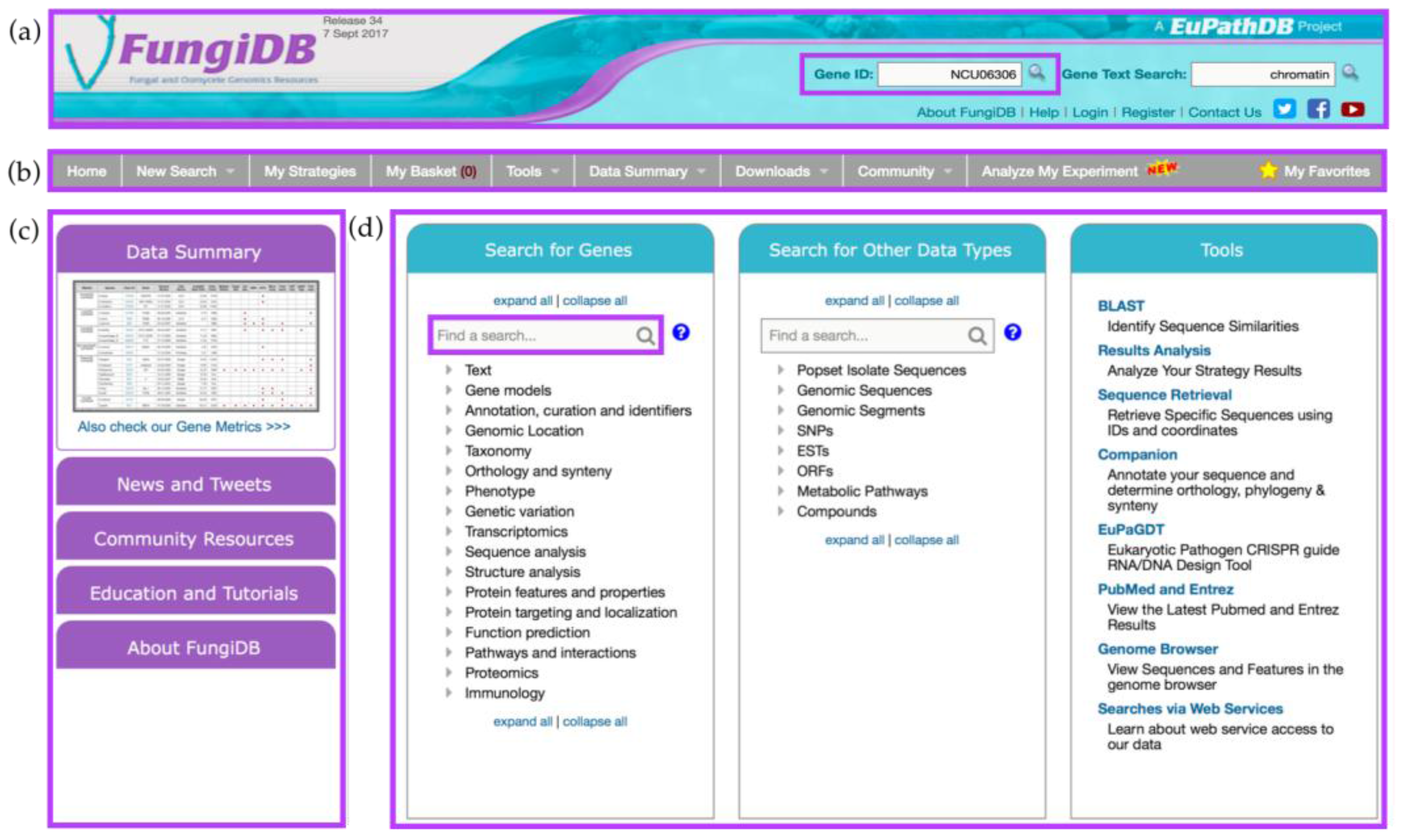
2. Overview of the FungiDB Home Page and its Features
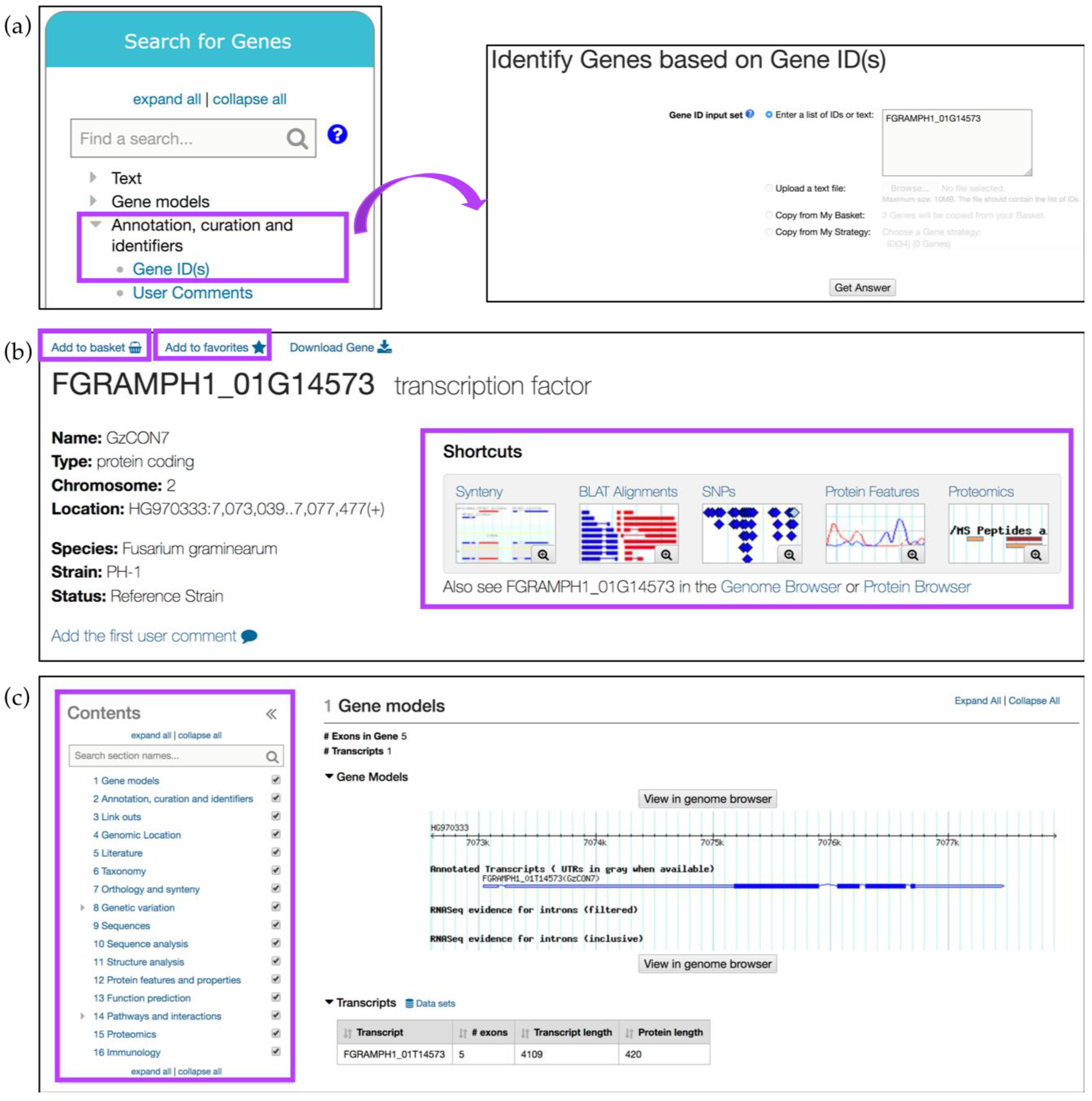
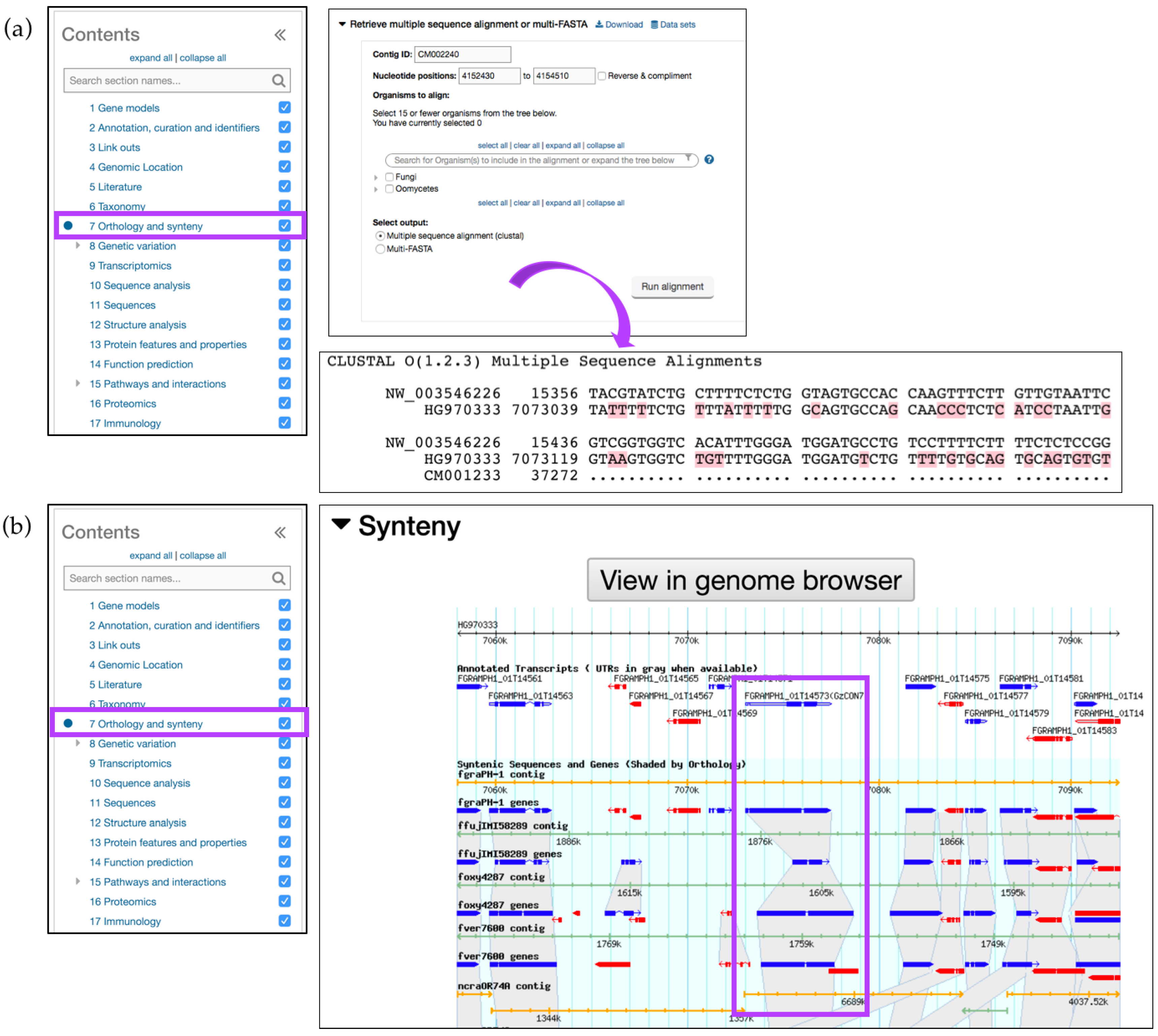
3. Exploring FungiDB Records: Gene ID Searches and Gene Record Pages
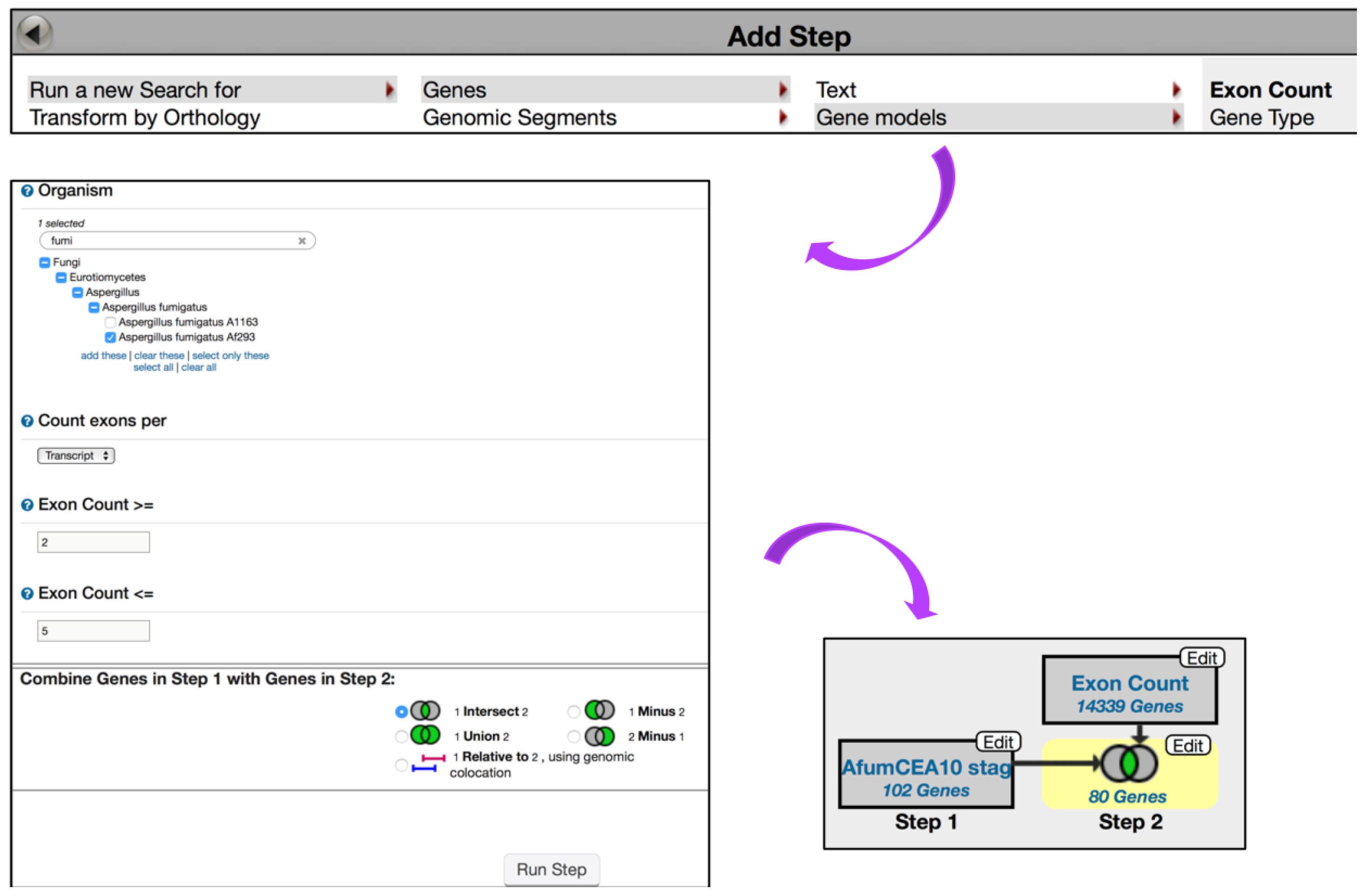
4. Creating in Silico Experiments via the FungiDB Query Interface
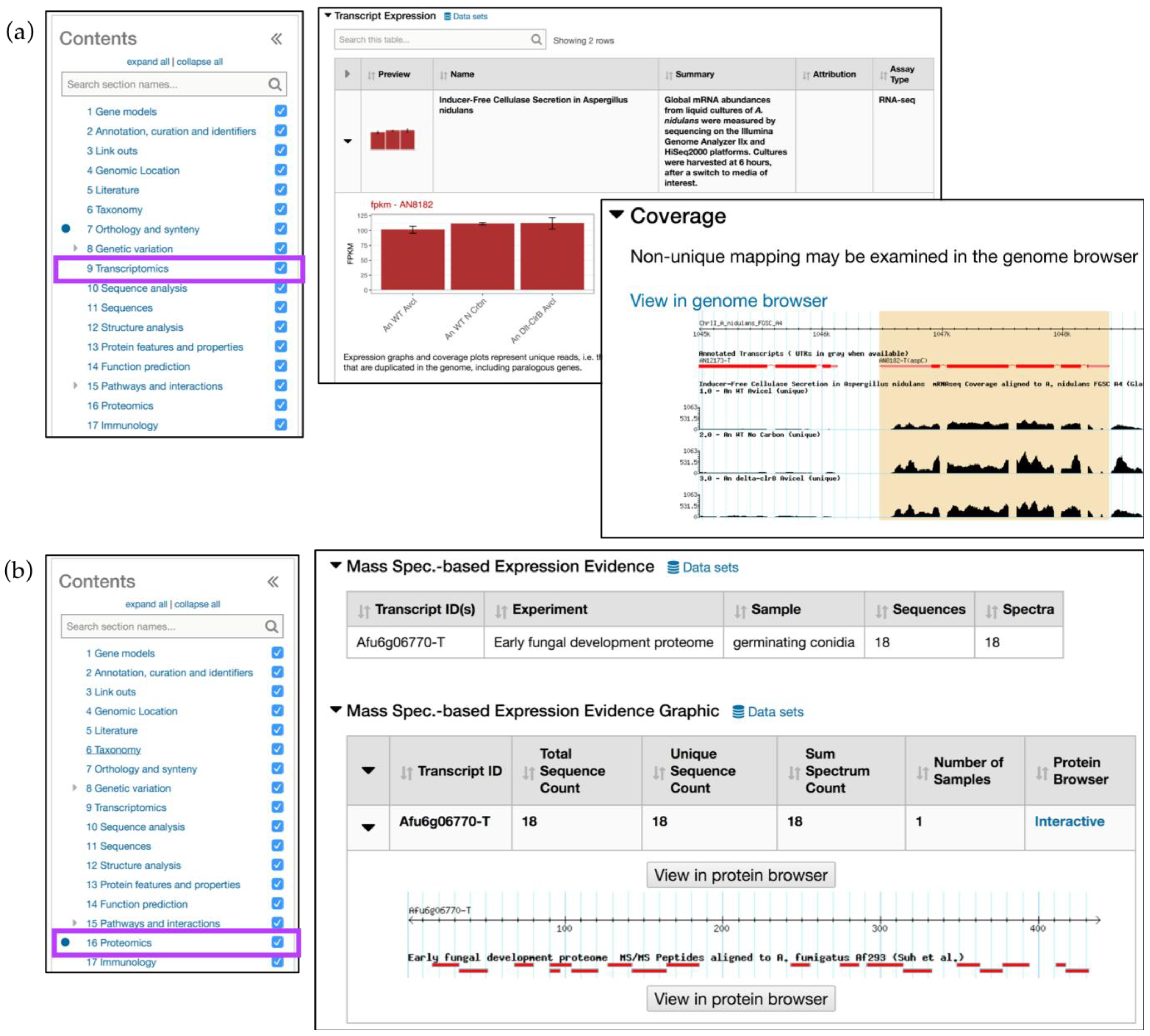
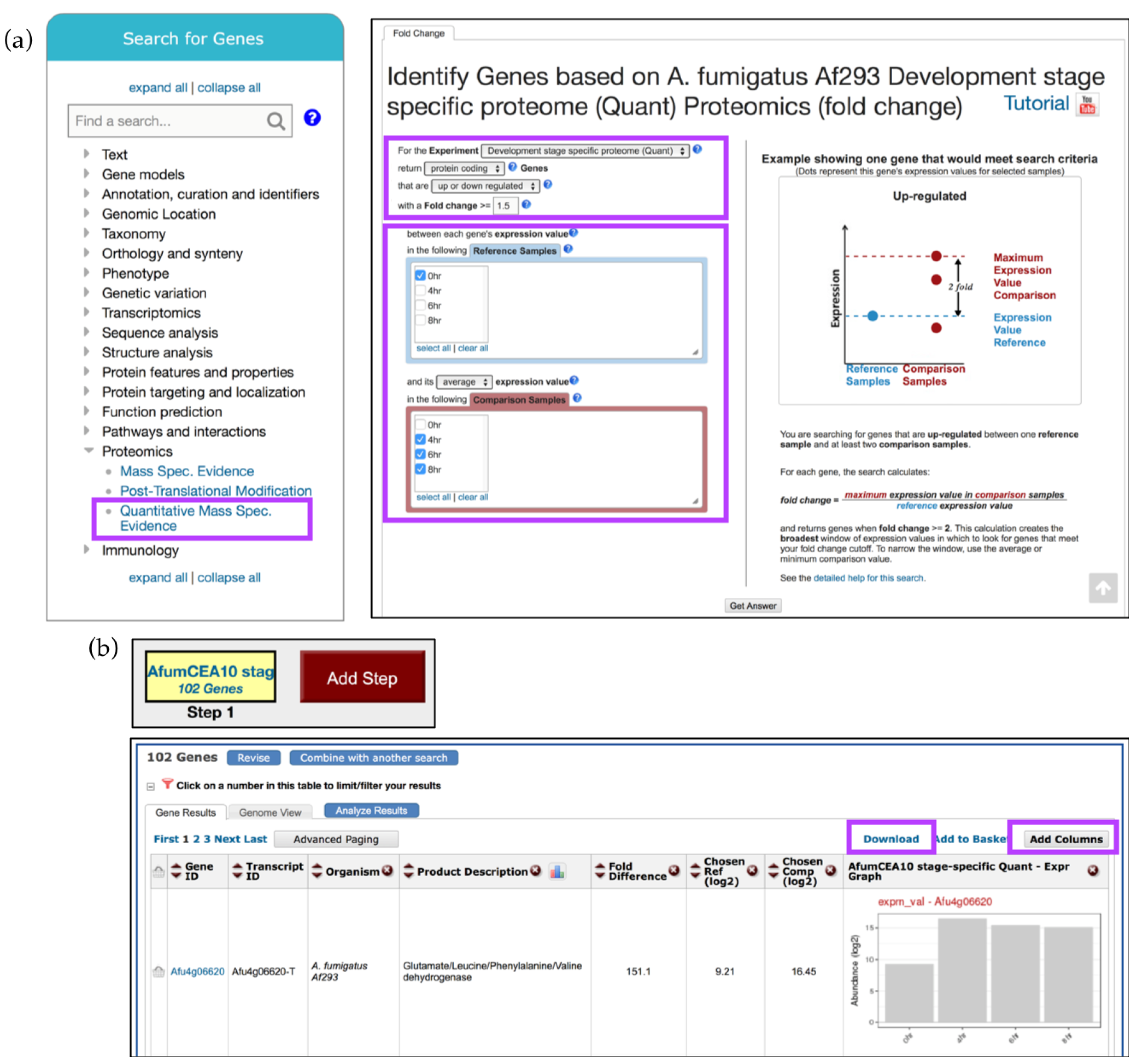
4.1. Mining Proteomic Datasets
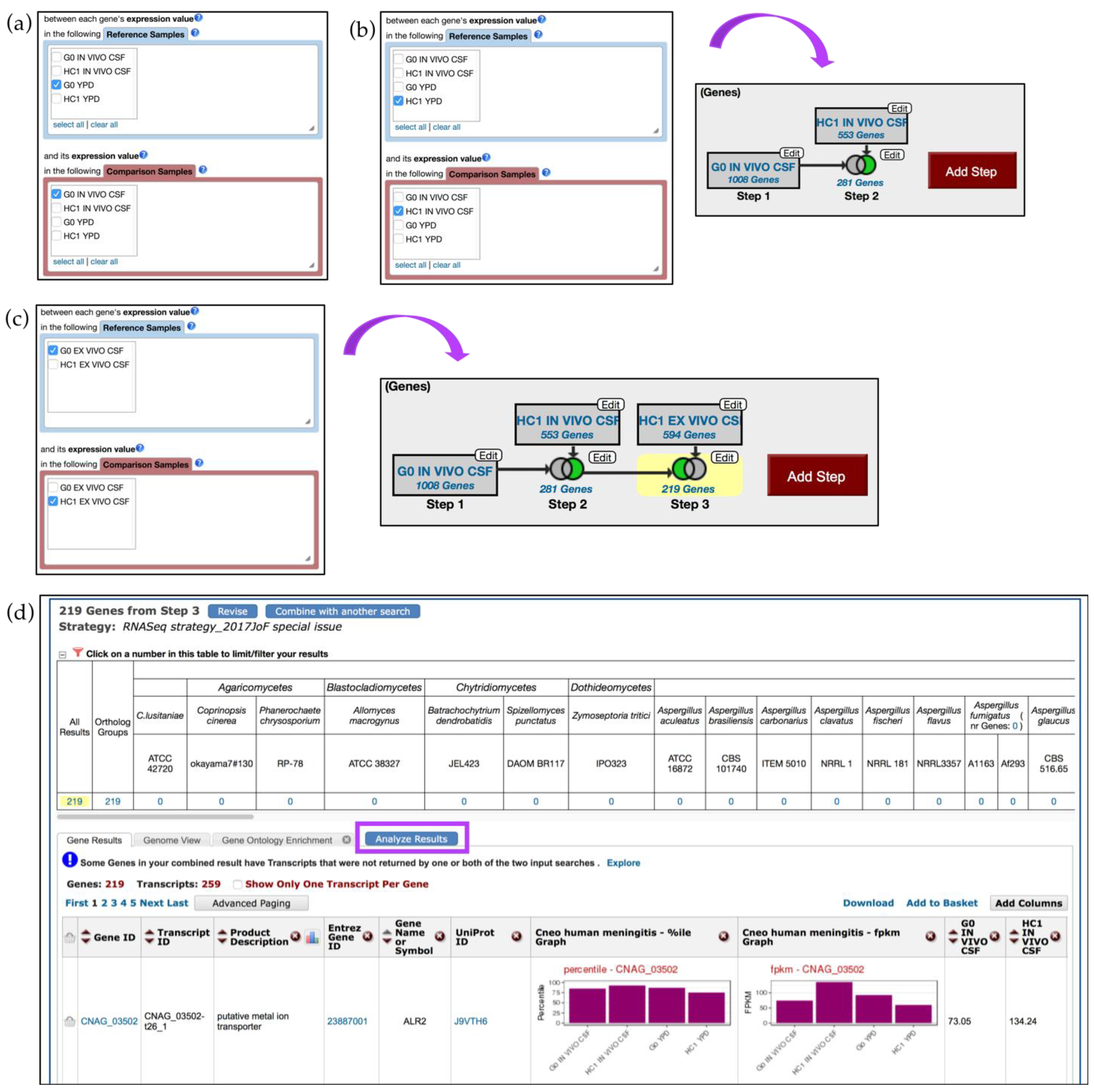
4.2. Creating In Silico RNA-Seq Experiments
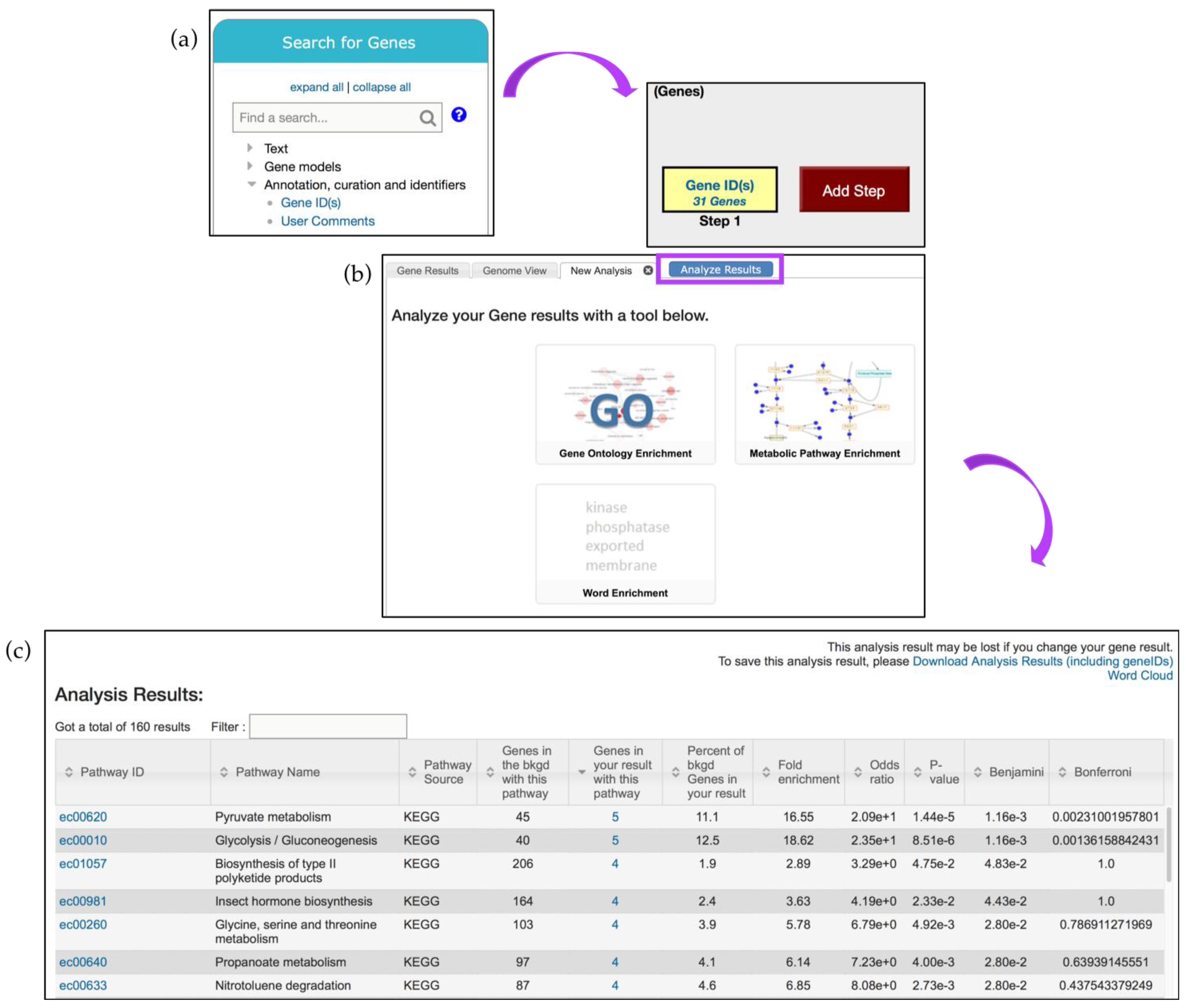
5. Additional Tools for Enrichment and Data Analysis
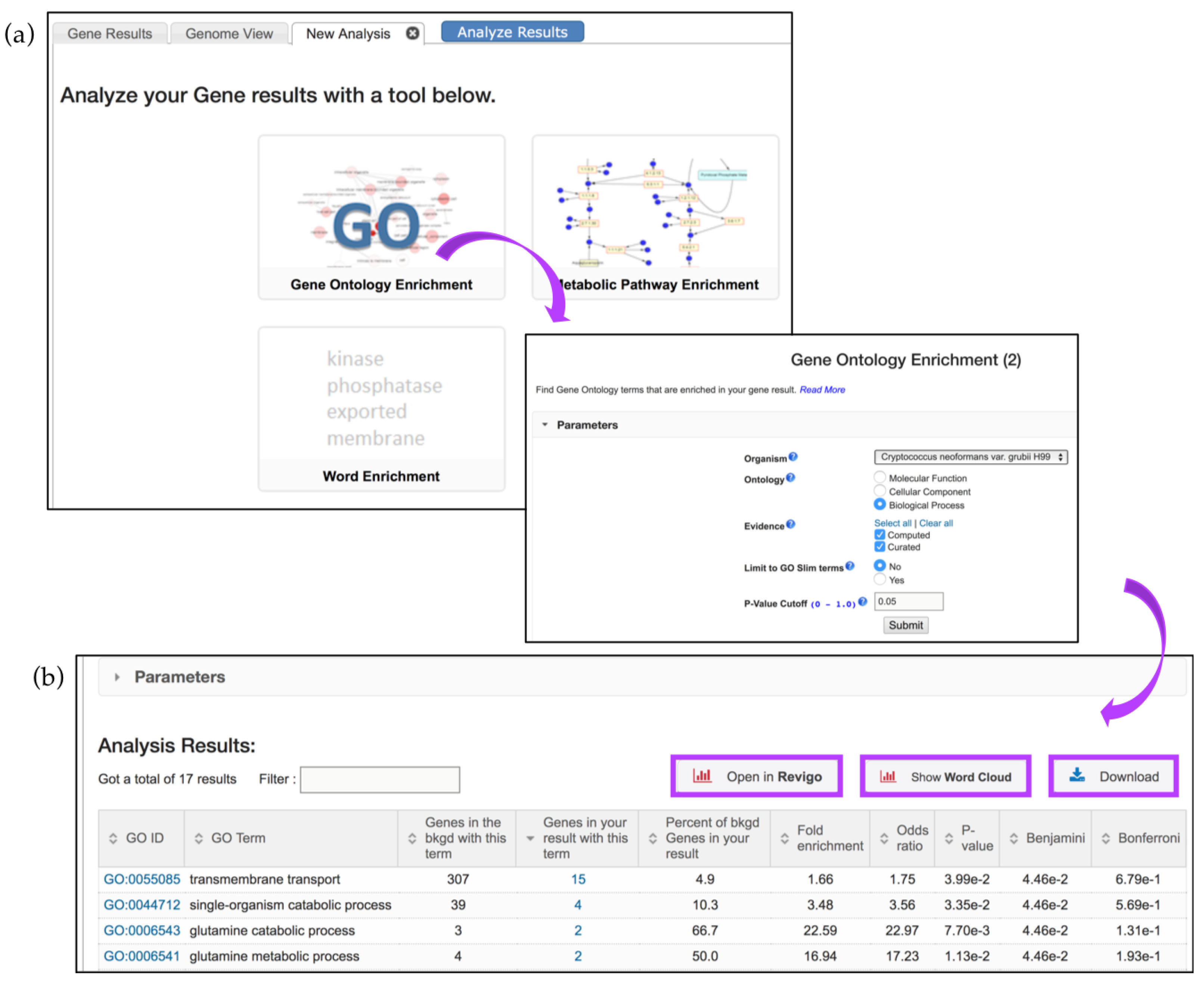
5.1. GO Term Enrichment and Visualization
- Fold enrichment—The ratio of the proportion of genes in the list of interest with a specific GO term over the proportion of genes in the background with that term;
- Odds ratio—Determines if the odds of the GO term appearing in the list of interest are the same as that for the background list;
- p-value—Assumptions under a null hypothesis, the probability of getting a result that is equal or greater than what was observed;
- Benjamini–Hochburg false discovery rate—A method for controlling false discovery rates for type 1 errors;
- Bonferroni-adjusted p-values—A method for correcting significance based on multiple comparisons;
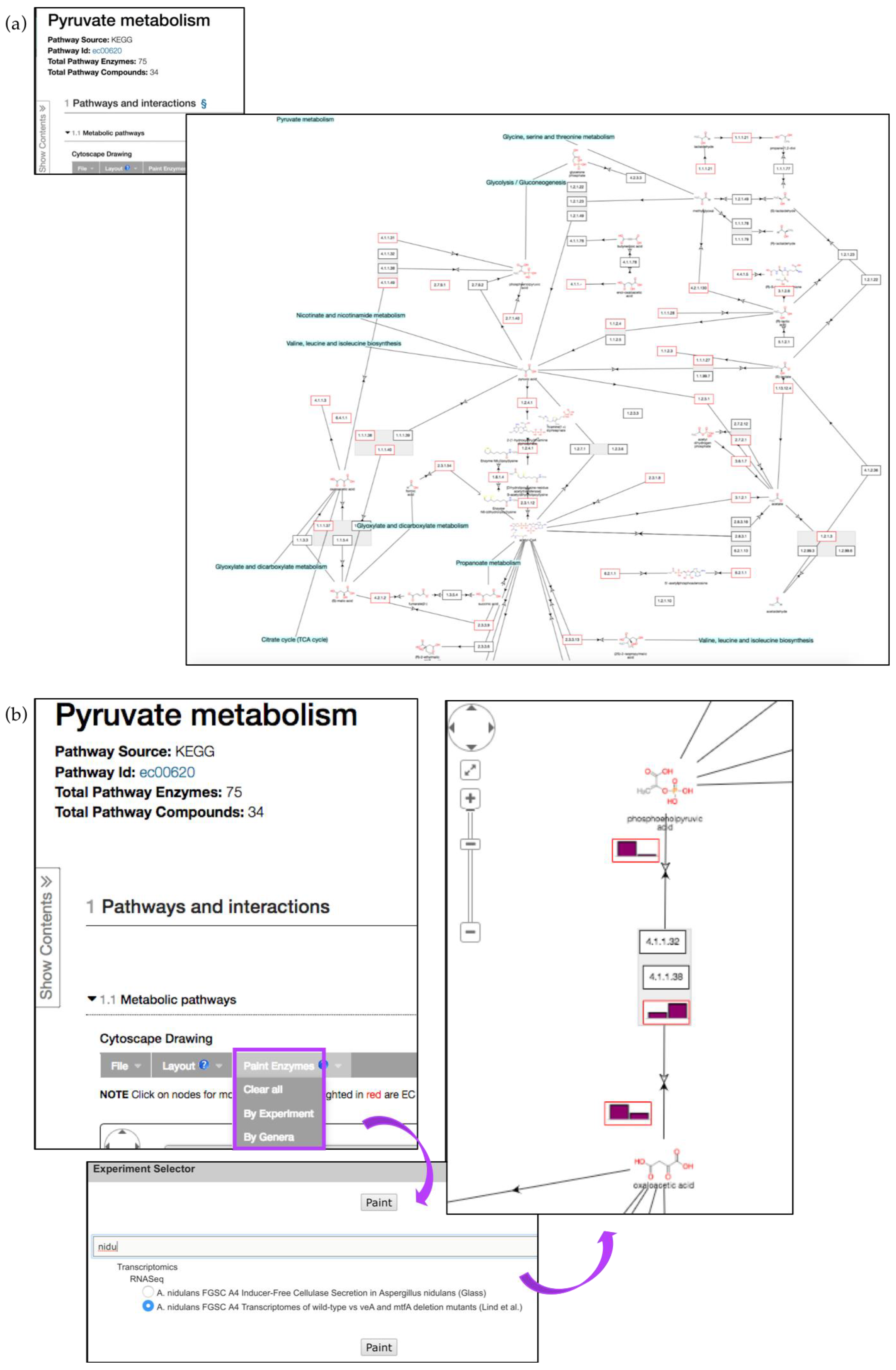
5.2. Metabolic Pathway Enrichment
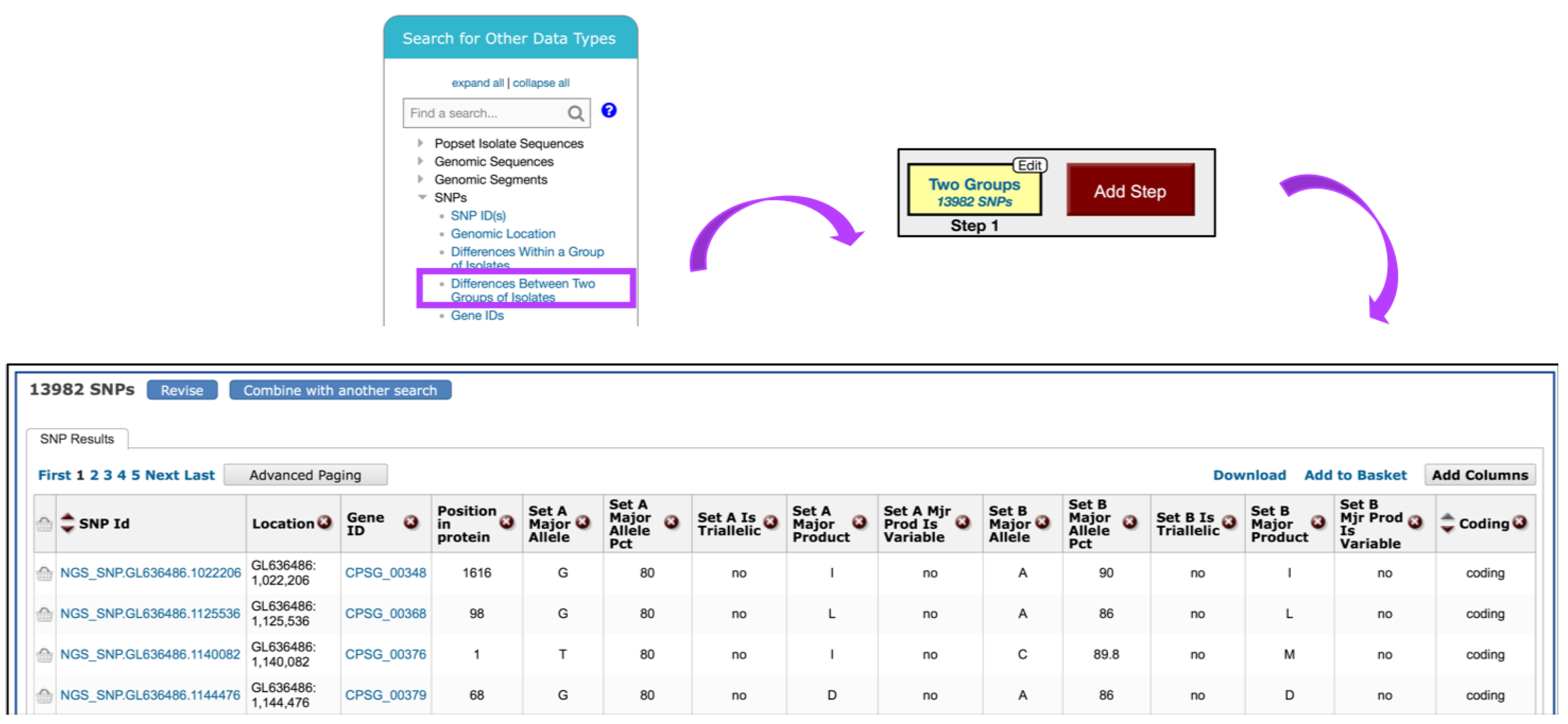
6. Data Mining via the Search for Other Data Types Panel
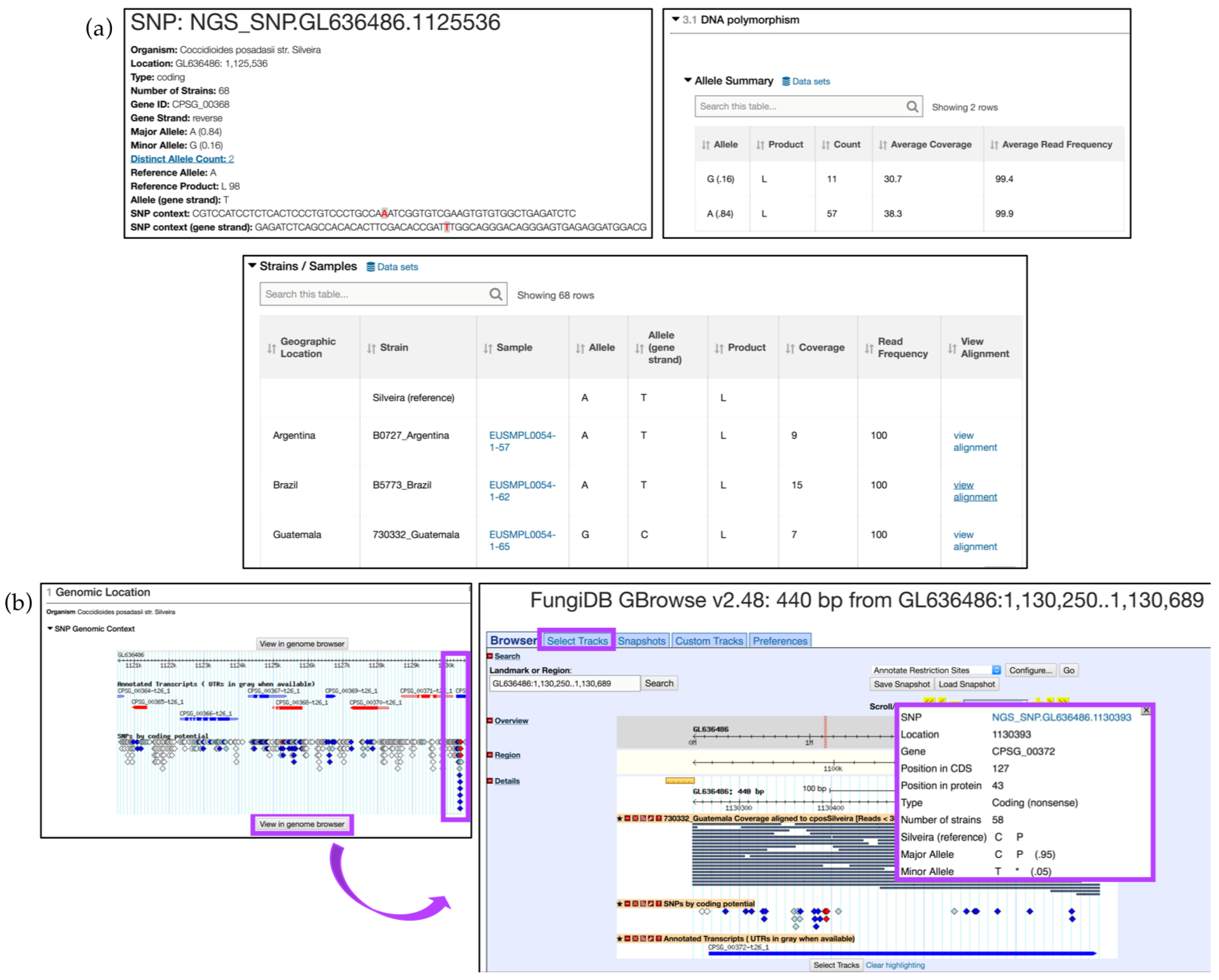
6.1. Identifying SNPs between Fungal Isolates Collected in Various Geographical Areas
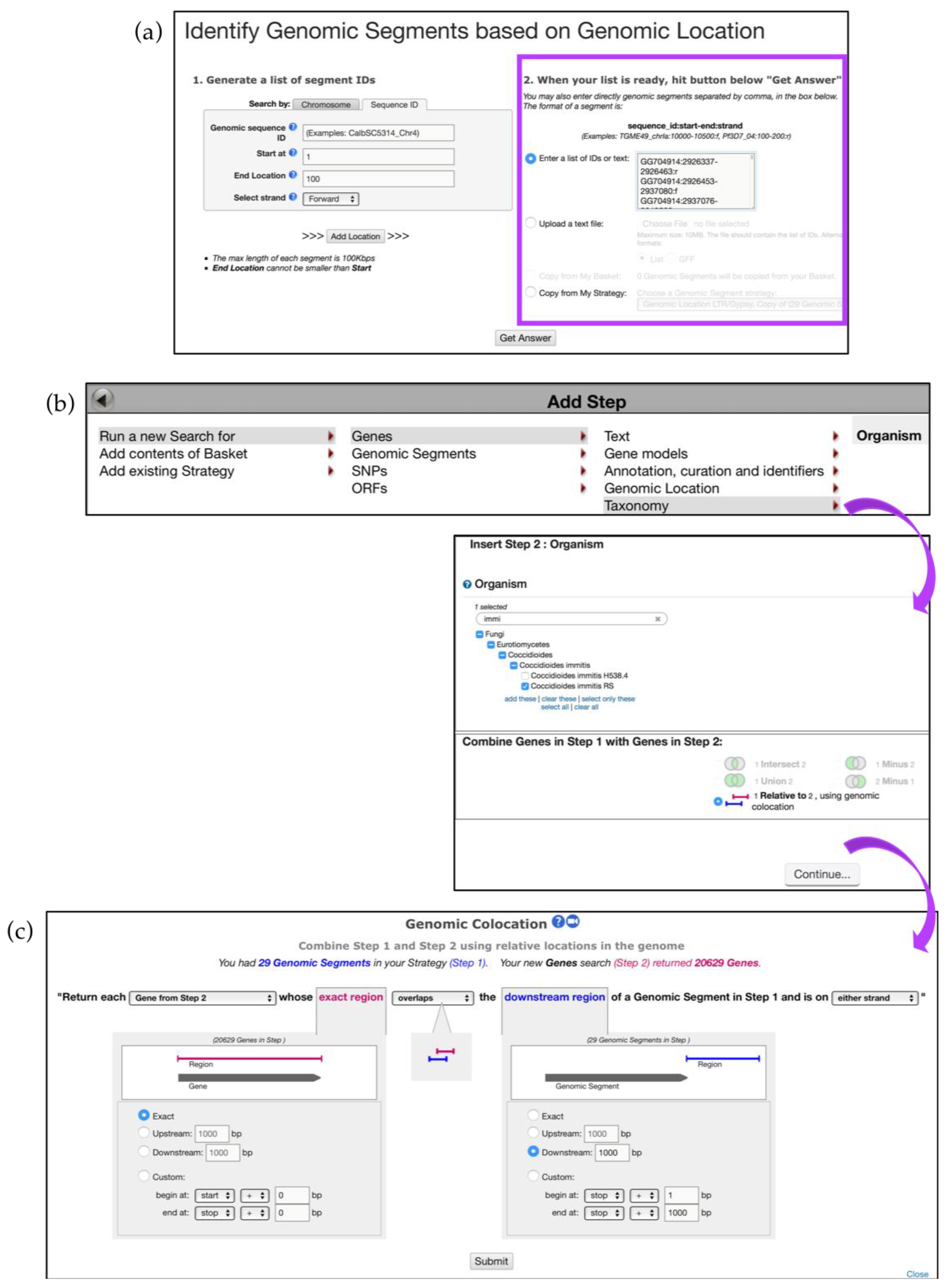
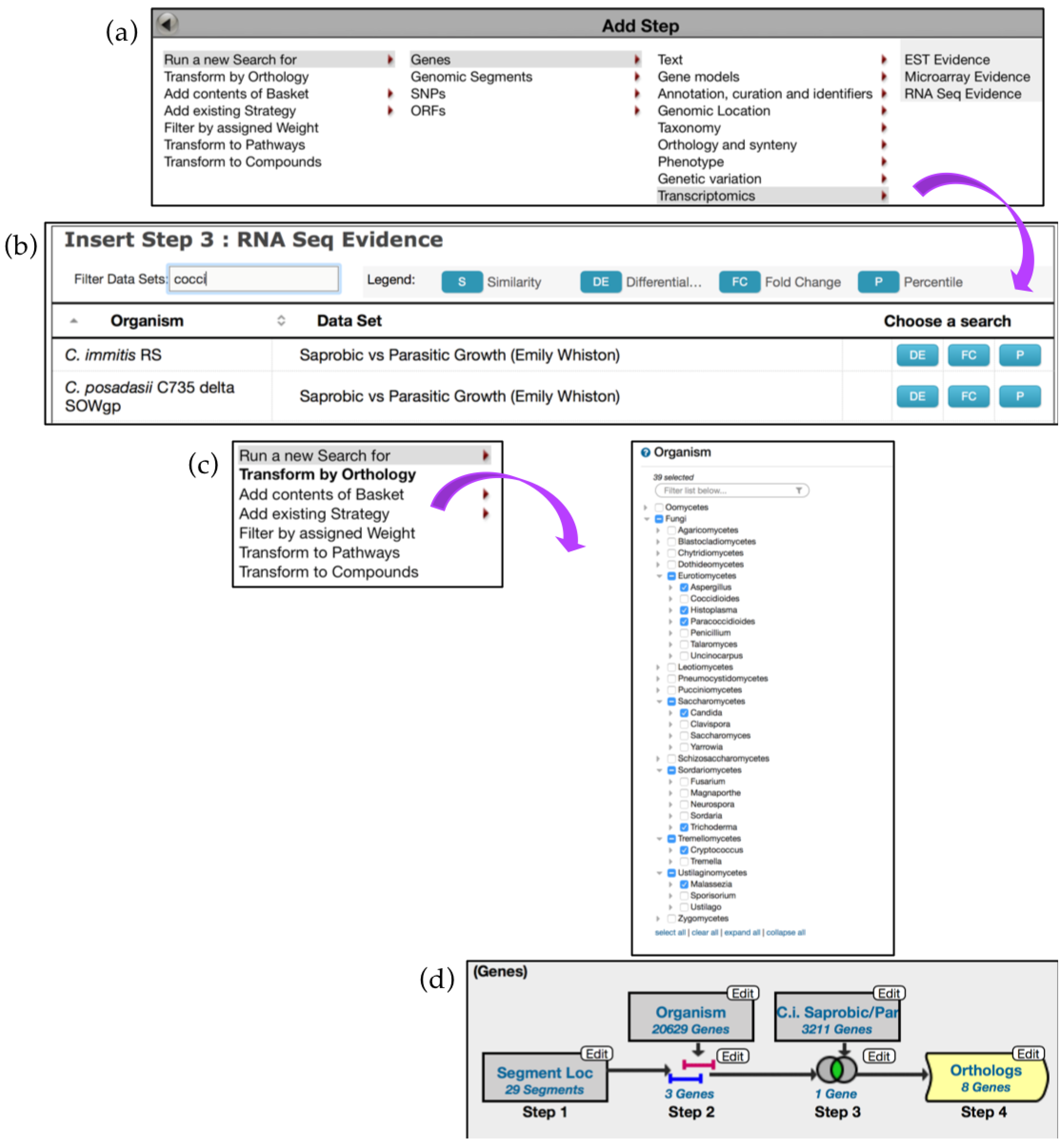
6.2. Utilizing the Genomic Location Search and Genomic Colocation Tool in Custom Queries
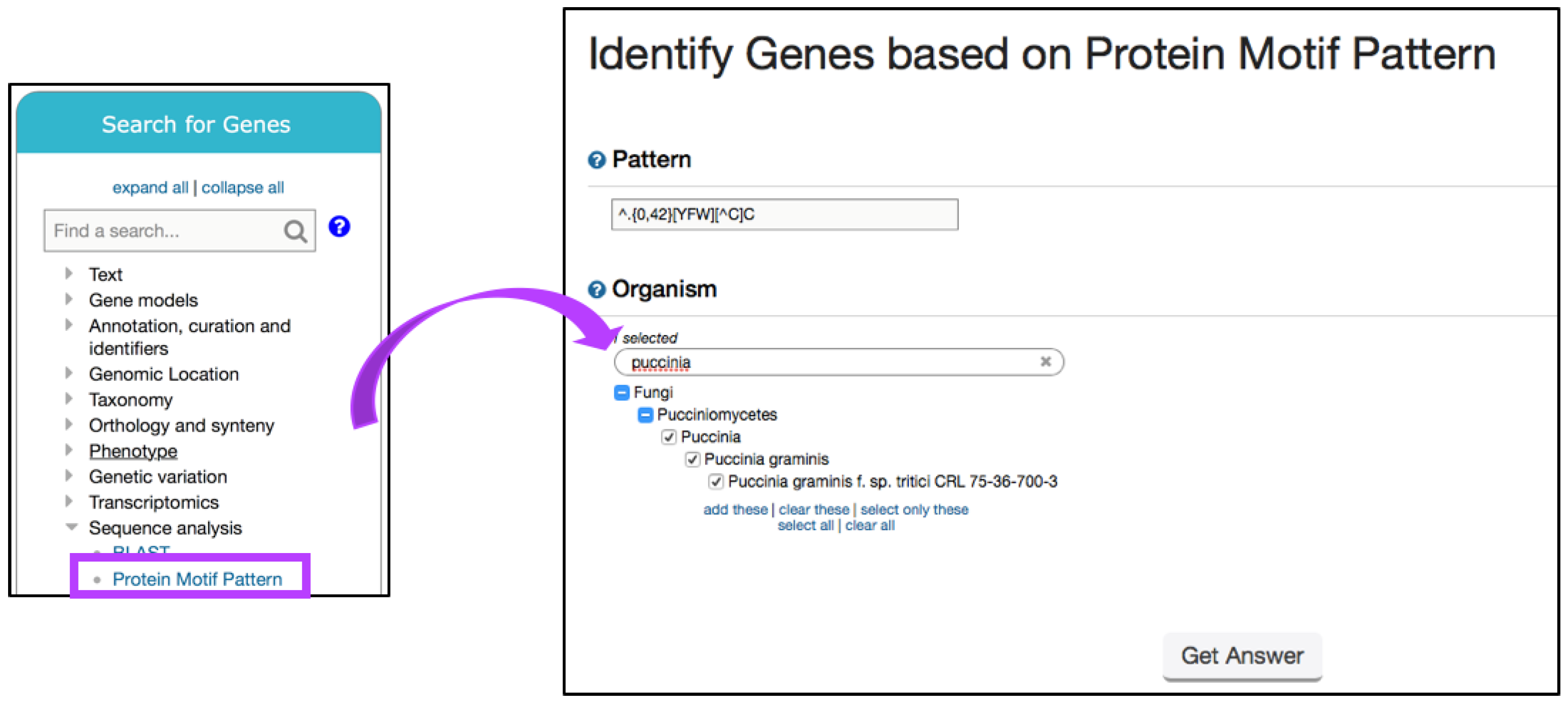
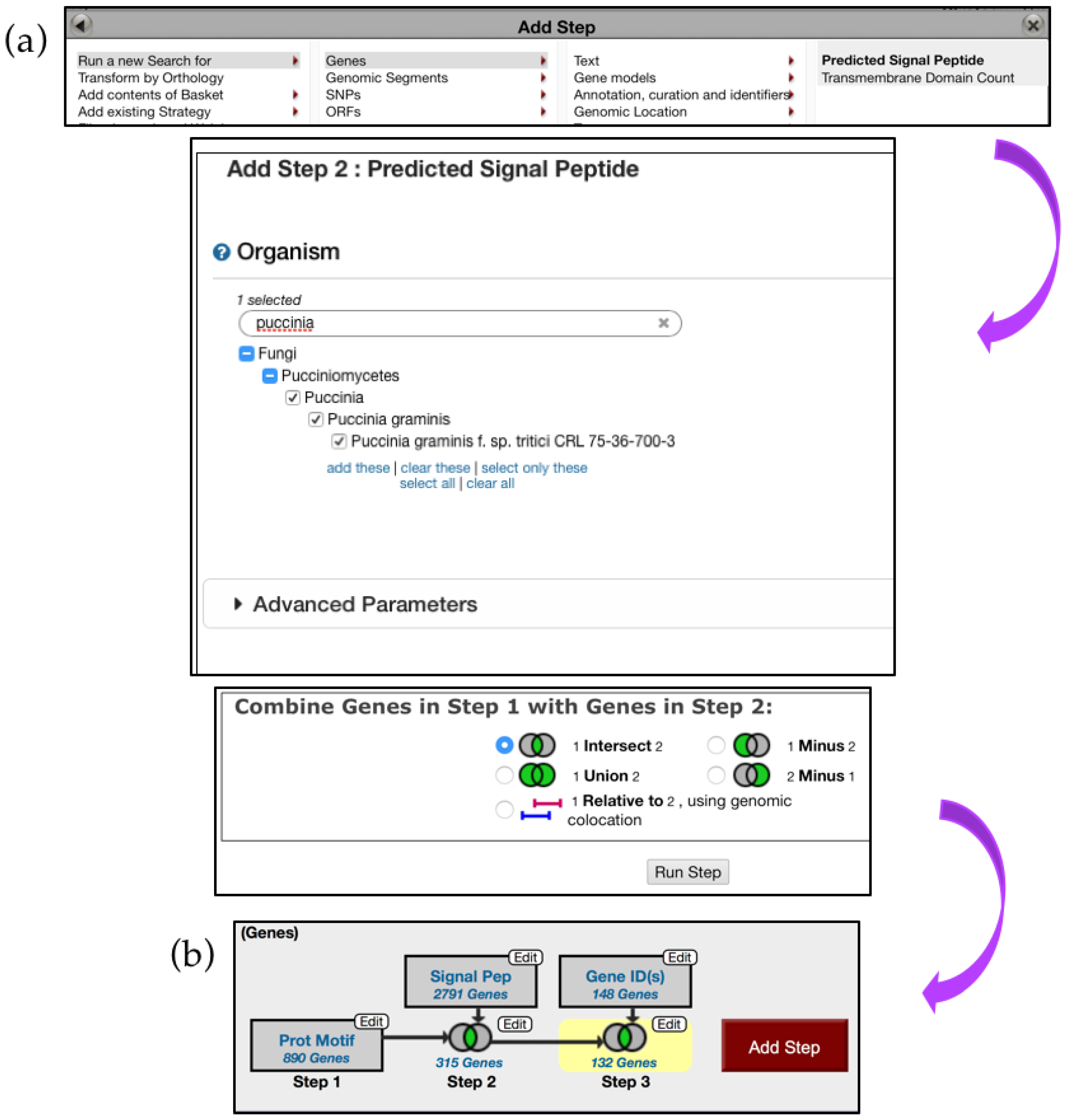
6.3. Searching for Fungal-Specific Motifs
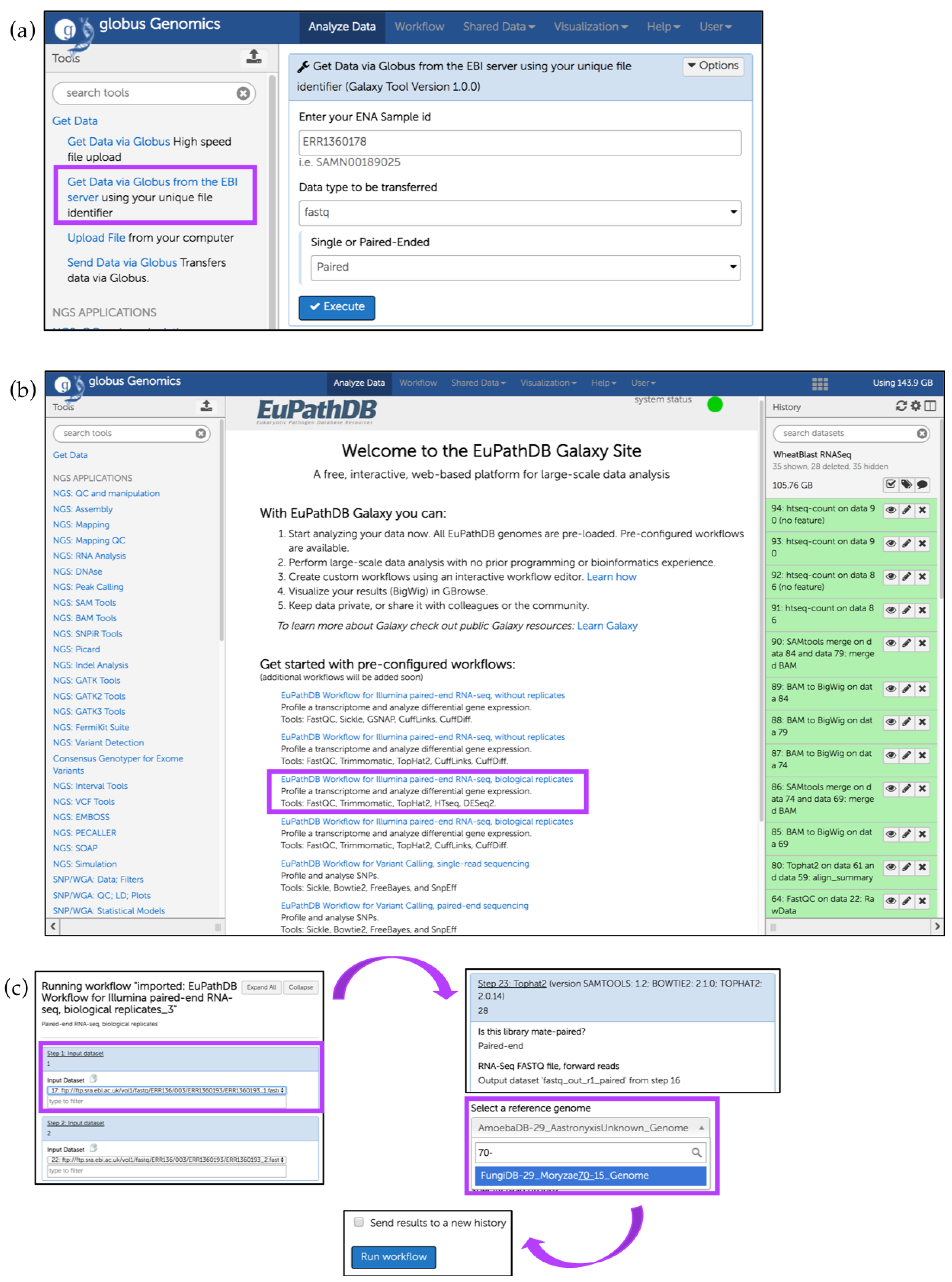
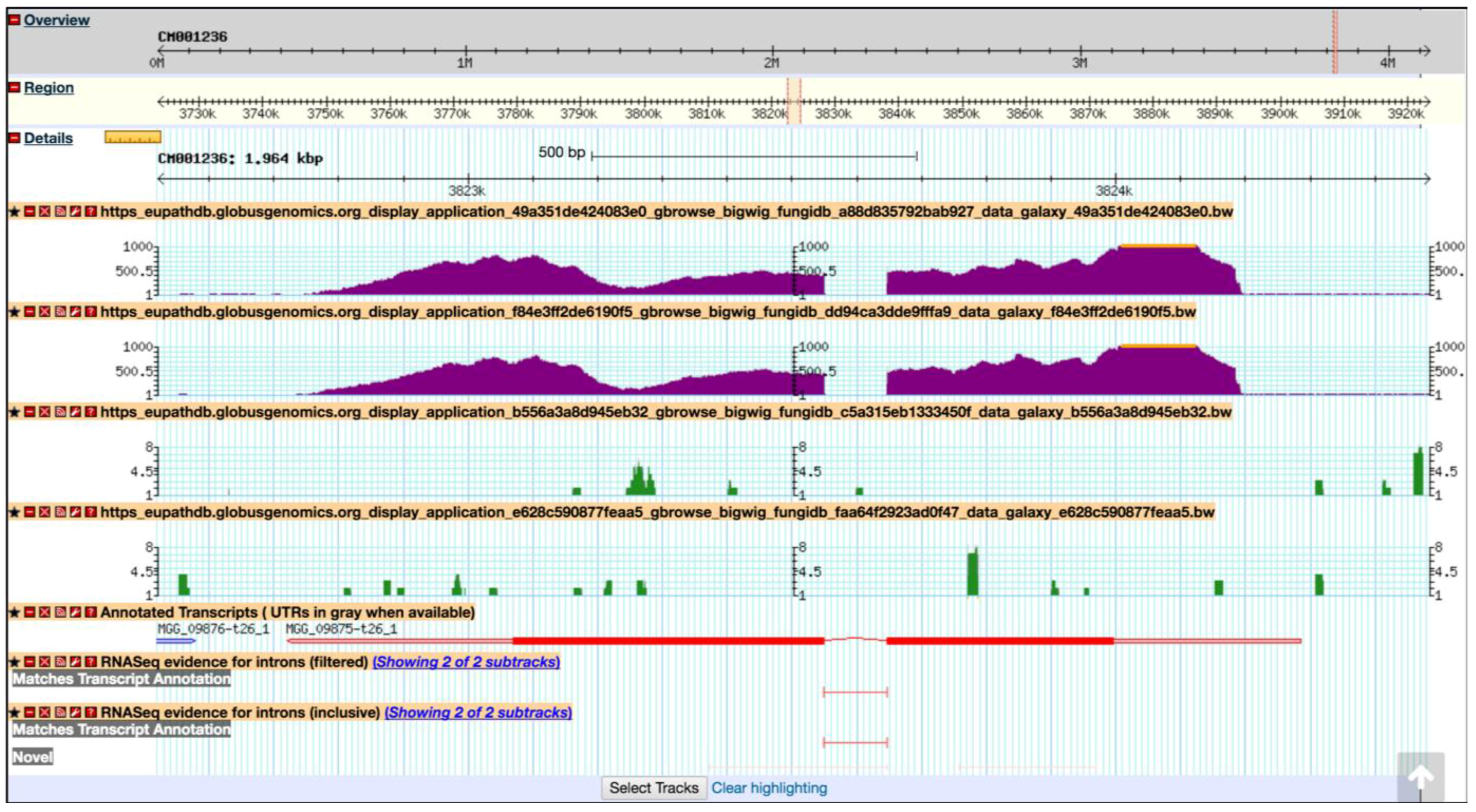
7. EuPathDB/FungiDB Galaxy Workspace
Running a Preconfigured (Sample) Workflow
8. Retrieve Sequences via Sequence Retrieval Tool
9. Empowering Users to Improve Genome Annotation
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stajich, J.E.; Harris, T.; Brunk, B.P.; Brestelli, J.; Fischer, S.; Harb, O.S.; Kissinger, J.C.; Li, W.; Nayak, V.; Pinney, D.F.; et al. FungiDB: An integrated functional genomics database for fungi. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- Aurrecoechea, C.; Barreto, A.; Basenko, E.Y.; Brestelli, J.; Brunk, B.P.; Cade, S.; Crouch, K.; Doherty, R.; Falke, D.; Fischer, S.; et al. EuPathDB: The eukaryotic pathogen genomics database resource. Nucleic Acids Res. 2017, 45, D581–D591. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.; Akanni, W.; Amode, M.R.; Barrell, D.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; Fitzgerald, S.; Gil, L.; et al. Ensembl 2016. Nucleic Acids Res. 2016, 44, D710–D716. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, H.; Cantor, M.; Dusheyko, S.; Hua, S.; Poliakov, A.; Shabalov, I.; Smirnova, T.; Grigoriev, I.V.; Dubchak, I. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, G.C.; Arnaud, M.B.; Inglis, D.O.; Skrzypek, M.S.; Binkley, G.; Simison, M.; Miyasato, S.R.; Binkley, J.; Orvis, J.; Shah, P.; et al. The Aspergillus Genome Database: Multispecies curation and incorporation of RNA-Seq data to improve structural gene annotations. Nucleic Acids Res. 2014, 42, D705–D710. [Google Scholar]
- Skrzypek, M.S.; Binkley, J.; Binkley, G.; Miyasato, S.R.; Simison, M.; Sherlock, G. The Candida Genome Database (CGD): Incorporation of Assembly 22, systematic identifiers and visualization of high throughput sequencing data. Nucleic Acids Res. 2017, 45, D592–D596. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces Genome Database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- McDowall, M.D.; Harris, M.A.; Lock, A.; Rutherford, K.; Staines, D.M.; Bähler, J.Ü.; Kersey, P.J.; Oliver, S.G.; Wood, V. PomBase 2015: Updates to the fission yeast database. Nucleic Acids Res. 2015, 43, D656–D661. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, C.A.; Birren, B.W. The fungal genome initiative and lessons learned from genome sequencing. Methods Enzymol. 2010, 470, 833–855. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Roldán, C.; Pareja-Jaime, Y.; González-Reyes, J.A.; G-Roncero, M.I. The transcription factor Con7-1 is a master regulator of morphogenesis and virulence in Fusarium oxysporum. Mol. Plant Microbe Interact. 2015, 28, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez gene: Gene-centered information at NCBI. Nucleic Acids Res. 2011, 39. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, K.; Wiest, A.; Plamann, M. The fungal genetics stock center: A repository for 50 years of fungal genetics research. J. Biosci. 2010, 35, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Winnenburg, R. PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 2006, 34, D459–D464. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Capella-Gutiérrez, S.; Pryszcz, L.P.; Marcet-Houben, M.; Gabaldón, T. PhylomeDB v4: Zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, T.T.; Basenko, E.; Harb, O.; Brown, N.A.; Urban, M.; Hammond-Kosack, K.E.; Bregitzer, P.P. Sharing mutants and experimental information prepublication using FgMutantDb (https://scabusa.org/FgMutantDb). Fungal Genet. Biol. 2018. [Google Scholar]
- Perkins, D.D.; Radford, A.; Sachs, M.S. The Neurospora Compendium; Academic Press: Cambridge, MA, USA, 2000; ISBN 0-12-550751-8. [Google Scholar]
- Dewey, C.N. Aligning multiple whole genomes with mercator and MAVID. Methods Mol. Biol. 2007, 395, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Coradetti, S.T.; Xiong, Y.; Glass, N.L. Analysis of a conserved cellulase transcriptional regulator reveals inducer-independent production of cellulolytic enzymes in Neurospora crassa. Microbiologyopen 2013, 2, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef] [PubMed]
- Suh, M.J.; Fedorova, N.D.; Cagas, S.E.; Hastings, S.; Fleischmann, R.D.; Peterson, S.N.; Perlin, D.S.; Nierman, W.C.; Pieper, R.; Momany, M. Development stage-specific proteomic profiling uncovers small, lineage specific proteins most abundant in the Aspergillus fumigatus conidial proteome. Proteome Sci. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Toffaletti, D.L.; Tenor, J.L.; Litvintseva, A.P.; Fang, C.; Mitchell, T.G.; McDonald, T.R.; Nielsen, K.; Boulware, D.R.; Bicanic, T.; et al. The Cryptococcus neoformans transcriptome at the site of human meningitis. MBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Molloy, S.F.; Chiller, T.; Greene, G.S.; Burry, J.; Govender, N.P.; Kanyama, C.; Mfinanga, S.; Lesikari, S.; Mapoure, Y.N.; Kouanfack, C.; et al. Cryptococcal meningitis: A neglected NTD? PLoS Negl. Trop. Dis. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. Expansion of the gene ontology knowledgebase and resources: The gene ontology consortium. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Simes, R.J. An improved bonferroni procedure for multiple tests of significance. Biometrika 1986, 73, 751–754. [Google Scholar] [CrossRef]
- Kolde, R.; Vilo, J. GOsummaries: an R Package for visual functional annotation of experimental data. F1000Research 2015. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Lima, P.; Casaletti, L.; Bailão, A.M.; de Vasconcelos, A.T.R.; da Rocha Fernandes, G.; de Almeida Soares, C.M. Transcriptional and proteomic responses to carbon starvation in Paracoccidioides. PLoS Negl. Trop. Dis. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Sharpton, T.J.; Stajich, J.E.; Rounsley, S.D.; Gardner, M.J.; Wortman, J.R.; Jordar, V.S.; Maiti, R.; Kodira, C.D.; Neafsey, D.E.; Zeng, Q.; et al. Comparative genomic analyses of the human fungal pathogens Coccidioides and their relatives. Genome Res. 2009, 19, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Noble, J.A.; Nelson, R.G.; Fufaa, G.D.; Kang, P.; Shafir, S.C.; Galgiani, J.N. Effect of geography on the analysis of coccidioidomycosis-associated deaths, United States. Emerg. Infect. Dis. 2016, 22, 1821–1823. [Google Scholar] [CrossRef] [PubMed]
- Engelthaler, D.M.; Roe, C.C.; Hepp, C.M.; Teixeira, M.; Driebe, E.M.; Schupp, J.M.; Gade, L.; Waddell, V.; Komatsu, K.; Arathoon, E.; et al. Local population structure and patterns of Western Hemisphere dispersal for Coccidioides spp., the fungal cause of Valley fever. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.; Böhlenius, H.; Pedersen, C.; Zhang, Z.; Emmersen, J.; Thordal-Christensen, H. Powdery mildew fungal effector candidates share N-terminal Y/F/WxC-motif. BMC Genom. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed]
- Madduri, R.K.; Sulakhe, D.; Lacinski, L.; Liu, B.; Rodriguez, A.; Chard, K.; Dave, U.J.; Foster, I.T. Experiences building Globus Genomics: A next-generation sequencing analysis service using Galaxy, Globus, and Amazon Web Services. Concurr. Comput. Pract. Exp. 2014, 26, 2266–2279. [Google Scholar] [CrossRef] [PubMed]
- Open Wheat Blast. Available online: http://s620715531.websitehome.co.uk/owb/ (accessed on 7 March 2018).
- Islam, M.T.; Croll, D.; Gladieux, P.; Soanes, D.M.; Persoons, A.; Bhattacharjee, P.; Hossain, M.S.; Gupta, D.R.; Rahman, M.M.; Mahboob, M.G.; et al. Emergence of wheat blast in Bangladesh was caused by a South American lineage of Magnaporthe oryzae. BMC Biol. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2017. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Mathioni, S.M.; Beló, A.; Rizzo, C.J.; Dean, R.A.; Donofrio, N.M. Transcriptome profiling of the rice blast fungus during invasive plant infection and in vitro stresses. BMC Genom. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Emmert, D.; Zhang, P. Using Chado to Store Genome Annotation Data. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002; ISBN 9780471250951. [Google Scholar]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Character | Meaning |
|---|---|
| A, B, C … | Amino acid single letter abbreviation for peptide motifs |
| [^a] | Any character except a |
| {n,m} | Match the preceding character between n and m times |
| [ ] | Match any character contained in the bracket |
| Comment type | Example Comment |
|---|---|
| Gene name, gene synonym | Cell division control protein Cdc48 is also known as dsc-6 |
| Functional characterization | This is a transcription factor involved in the regulation of copper import |
| Subcellular localization | GFP tagging demonstrates that ham-5 co-localize with mak-2. Images attached |
| Phenotype | Deletion of rim101 has resulted in capsule defects and increased virulence in mice models |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basenko, E.Y.; Pulman, J.A.; Shanmugasundram, A.; Harb, O.S.; Crouch, K.; Starns, D.; Warrenfeltz, S.; Aurrecoechea, C.; Stoeckert, C.J., Jr.; Kissinger, J.C.; et al. FungiDB: An Integrated Bioinformatic Resource for Fungi and Oomycetes. J. Fungi 2018, 4, 39. https://doi.org/10.3390/jof4010039
Basenko EY, Pulman JA, Shanmugasundram A, Harb OS, Crouch K, Starns D, Warrenfeltz S, Aurrecoechea C, Stoeckert CJ Jr., Kissinger JC, et al. FungiDB: An Integrated Bioinformatic Resource for Fungi and Oomycetes. Journal of Fungi. 2018; 4(1):39. https://doi.org/10.3390/jof4010039
Chicago/Turabian StyleBasenko, Evelina Y., Jane A. Pulman, Achchuthan Shanmugasundram, Omar S. Harb, Kathryn Crouch, David Starns, Susanne Warrenfeltz, Cristina Aurrecoechea, Christian J. Stoeckert, Jr., Jessica C. Kissinger, and et al. 2018. "FungiDB: An Integrated Bioinformatic Resource for Fungi and Oomycetes" Journal of Fungi 4, no. 1: 39. https://doi.org/10.3390/jof4010039
APA StyleBasenko, E. Y., Pulman, J. A., Shanmugasundram, A., Harb, O. S., Crouch, K., Starns, D., Warrenfeltz, S., Aurrecoechea, C., Stoeckert, C. J., Jr., Kissinger, J. C., Roos, D. S., & Hertz-Fowler, C. (2018). FungiDB: An Integrated Bioinformatic Resource for Fungi and Oomycetes. Journal of Fungi, 4(1), 39. https://doi.org/10.3390/jof4010039