Seven New Cytotoxic and Antimicrobial Xanthoquinodins from Jugulospora vestita

 ,
,  and
and 
Abstract
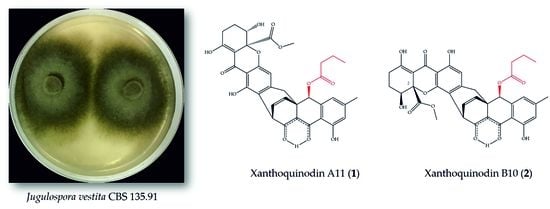
1. Introduction
2. Materials and Methods
2.1. General
2.2. Fermentation, Extraction, and Isolation
2.3. Spectral Data
2.3.1. Xanthoquinodin A11 (1)
2.3.2. Xanthoquinodin B10 (2)
2.3.3. Xanthoquinodin B11 (3)
2.3.4. Xanthoquinodin B12 (4)
2.3.5. Xanthoquinodin B13 (5)
2.3.6. Xanthoquinodin B14 (6)
2.3.7. Xanthoquinodin B15 (7)
2.3.8. Xanthoquinodin B4 (8)
2.4. Biological Assays
2.5. Phylogenetic Study
3. Results and Discussion
3.1. Structure Elucidation of Compounds 1–7
3.2. Antimicrobial and Cytotoxic Activities of Compounds 1–8
3.3. Comparison of Secondary Metabolite Production of Jugulospora spp.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cooper, M.A.; Shlaes, D. Fix the antibiotics pipeline. Nature 2011, 472, 32. [Google Scholar] [CrossRef] [PubMed]
- Sandargo, B.; Chepkirui, C.; Cheng, T.; Chaverra-Munoz, L.; Thongbai, B.; Stadler, M.; Hüttel, S. Biological and chemical diversity go hand in hand: Basidiomycota as source of new pharmaceuticals and agrochemicals. Biotechnol. Adv. 2019, 37, 107344. [Google Scholar] [CrossRef]
- Rupcic, Z.; Rascher, M.; Kanaki, S.; Köster, R.W.; Stadler, M.; Wittstein, K. Two new cyathane diterpenoids from mycelial cultures of the medicinal mushroom Hericium erinaceus and the rare species, Hericium flagellum. Int. J. Mol. Sci. 2018, 19, 740. [Google Scholar] [CrossRef]
- Padma, V.V. An overview of targeted cancer therapy. BioMedicine 2015, 5, 19. [Google Scholar] [CrossRef]
- Alexandre, J.; Raymond, E.; Kaci, M.O.; Brain, E.C.; Lokiec, F.; Kahatt, C.; Faivre, S.; Yovine, A.; Goldwasser, F.; Smith, S.L.; et al. Phase I and pharmacokinetic study of irofulven administered weekly or biweekly in advanced solid tumor patients. Clin. Cancer Res. 2004, 10, 3377–3385. [Google Scholar] [CrossRef][Green Version]
- Miyamoto, M.; Takano, M.; Kuwahara, M.; Soyama, H.; Kato, K.; Matuura, H.; Sakamoto, T.; Takasaki, K.; Aoyama, T.; Yoshikawa, T.; et al. Efficacy of combination chemotherapy using irinotecan and nedaplatin for patients with recurrent and refractory endometrial carcinomas: Preliminary analysis and literature review. Cancer Chemother. Pharmacol. 2018, 81, 111–117. [Google Scholar] [CrossRef]
- Topka, S.; Khalil, S.; Stanchina, E.; Vijai, J.; Offit, K. Preclinical evaluation of enhanced irofulven antitumor activity in an ERCC3 mutant background by in vitro and in vivo tumor models. AACR 2018, 78, 3258. [Google Scholar]
- Surup, F.; Kuhnert, E.; Lehmann, E.; Heitkämper, S.; Hyde, K.D.; Fournier, J.; Stadler, M. Sporothriolide derivatives as chemotaxonomic markers for Hypoxylon monticulosum. Mycology 2014, 5, 110–119. [Google Scholar] [CrossRef]
- Helaly, S.E.; Thongbai, B.; Stadler, M. Diversity of biologically active secondary metabolites from endophytic and saprotrophic fungi of the ascomycete order Xylariales. Nat. Prod. Rep. 2018, 35, 992–1014. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Mohr, K.I.; Stadler, M.; Dickschat, J.S. Volatiles from the tropical ascomycete Daldinia clavata (Hypoxylaceae, Xylariales). Beilstein J. Org. Chem. 2018, 14, 135–147. [Google Scholar] [PubMed]
- Bills, G.; Gloer, J. Biologically active secondary metabolites from the fungi. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Vicente, F.; Basilio, A.; Platas, G.; Collado, J.; Bills, G.F.; González del Val, A.; Martín, J.; Tormo, J.R.; Harris, G.H.; Zink, D.L.; et al. Distribution of the antifungal agents sordarins across filamentous fungi. Mycol. Res. 2009, 113, 754–770. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, S.; Horie, Y. Two new species of terrestrial Ascomycetes from Eastern Nepal. In Reports on the Cryptogamic Study in Nepal; Otani, Y., Ed.; Miscellaneous Publication of the National Science Museum: Tokyo, Japan, 1982. [Google Scholar]
- Marin-Felix, Y.; Miller, A.N.; Cano-Lira, J.F.; Guarro, J.; García, D.; Stadler, M.; Huhndorf, S.M.; Stchigel, A.M. Re-evaluation of the order Sordariales: Delimitation of Lasiosphaeriaceae s. str., and introduction of the new families Diplogelasinosporaceae, Naviculisporaceae and Schizotheciaceae. Microorganisms 2020, 8, 1430. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Wessel, A.C.; Luangsa-ard, J.J.; Stadler, M. Viridistratins A-C, antimicrobial and cytotoxic benzo[j]fluoranthenes from stromata of Annulohypoxylon viridistratum (Hypoxylaceae, Ascomycota). Biomolecules 2020, 10, 805. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software v. 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 12, 2725–2729. [Google Scholar] [CrossRef]
- Mason-Gamer, R.; Kellogg, E. Testing for phylogenetic conflict among molecular data sets in the tribe Triticeae (Gramineae). Syst. Biol. 1996, 45, 524–545. [Google Scholar] [CrossRef]
- Wiens, J.J. Testing phylogenetic methods with tree congruence: Phylogenetic analysis of polymorphic morphological characters in phrynosomatid lizards. Syst. Biol. 1998, 47, 427–444. [Google Scholar] [CrossRef]
- Hernández-Restrepo, M.; Groenewald, J.Z.; Elliott, M.L.; Canning, G.; McMillan, V.E.; Crous, P.W. Take-all or nothing. Stud. Mycol. 2016, 83, 19–48. [Google Scholar] [CrossRef]
- Alfaro, M.E.; Zoller, S.; Lutzoni, F. Bayes or bootstrap. A simulation study comparing the performance of Bayesian Markov chainMonte Carlo sampling and bootstrapping in assessing phylogenetic confidence. Mol. Biol. Evol. 2003, 20, 255–266. [Google Scholar] [CrossRef]
- Chen, G.D.; Chen, Y.; Gao, H.; Shen, L.Q.; Wu, Y.; Li, X.X.; Li, Y.; Guo, L.D.; Cen, Y.Z.; Yao, X.S. Xanthoquinodins from the endolichenic fungal strain Chaetomium elatum. J. Nat. Prod. 2013, 76, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Sadorn, K.; Saepua, S.; Boonyuen, N.; Boonruangprapa, T.; Rachtawee, P.; Pittayakhajonwut, P. Antimicrobial activity and cytotoxicity of xanthoquinodin analogs from the fungus Cytospora eugeniae BCC42696. Phytochemistry 2018, 151, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Tantapakul, C.; Promgool, T.; Kanokmedhakul, K.; Soytong, K.; Song, J.; Hadsadee, S.; Jungsuttiwong, S.; Kanokmedhakul, S. Bioactive xanthoquinodins and epipolythiodioxopiperazines from Chaetomium globosum 7s-1, an endophytic fungus isolated from Rhapis cochinchinensis (Lour.) Mart. Nat. Prod. Res. 2018, 34, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Anaya-Eugenio, G.D.; Rebollar-Ramos, D.; Gonzalez, M.D.C.; Raja, H.; Mata, R.; Carcache de Blanco, E.J. Apoptotic activity of xanthoquinodin JBIR-99, from Parengyodontium album MEXU 30054, in PC-3 human prostate cancer cells. Chem. Biol. Interact. 2019, 311, 108798. [Google Scholar] [CrossRef] [PubMed]
- Ueda, J.Y.; Takagi, M.; Shin-ya, K. New xanthoquinodin-like compounds, JBIR-97, -98 and -99, obtained from marine sponge-derived fungus Tritirachium sp. SpB081112MEf2. J. Antibiot. 2010, 63, 615–618. [Google Scholar] [CrossRef]
- Tabata, N.; Tomoda, H.; Matsuzaki, K.; Omura, S. Structure and biosynthesis of xanthoquinodins, anticoccidial antibiotics. J. Am. Chem. Soc. 1993, 115, 8558–8564. [Google Scholar] [CrossRef]
- Tabata, N.; Suzumura, Y.; Tomoda, H.; Masuma, R.; Haneda, K.; Kishi, M.; Iwai, Y.; Omura, S. Xanthoquinodins, new anticoccidial agents produced by Humicola sp. J. Antibiot. 1993, 46, 749–755. [Google Scholar] [CrossRef]
- Tabata, N.; Tomoda, H.; Iwai, Y.; Omura, S. Xanthoquinodin B3, a new anticoccidal agent produced by Humicola sp. FO-888. J. Antibiot. 1995, 49, 267–271. [Google Scholar] [CrossRef]
- Wang, X.W.; Bai, F.Y.; Bensch, K.; Meijer, M.; Sun, B.D.; Han, Y.F.; Crous, P.W.; Samson, R.A.; Yang, F.Y.; Houbraken, J. Phylogenetic re-evaluation of Thielavia with the introduction of a new family Podosporaceae. Stud. Mycol. 2019, 93, 155–252. [Google Scholar] [CrossRef]
- Miller, A.N.; Huhndorf, S.M. A natural classification of Lasiosphaeria based on nuclear LSU rDNA sequences. Mycol. Res. 2004, 108, 26–34. [Google Scholar] [CrossRef]
- Miller, A.N.; Huhndorf, S. Multi-gene phylogenies indicate ascomal wall morphology is a better predictor of phylogenetic relationships than ascospore morphology in the Sordariales (Ascomycota, Fungi). Mol. Phylogenetics Evol. 2005, 35, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Sir, E.B.; Kuhnert, E.; Lambert, C.; Hladki, A.I.; Romero, A.I.; Stadler, M. New species and reports of Hypoxylon from Argentina recognized by a polyphasic approach. Mycol. Prog. 2016, 15, 42. [Google Scholar] [CrossRef]
- Kuhnert, E.; Sir, E.B.; Lambert, C.; Hyde, K.D.; Hladki, A.I.; Romero, A.I.; Rohde, M.; Stadler, M. Phylogenetic and chemotaxonomic resolution of the genus Annulohypoxylon (Xylariaceae) including four new species. Fungal Divers. 2017, 85, 1–43. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | |||||
|---|---|---|---|---|---|---|---|---|
| No | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) |
| 2 | 84.3, C | – | 85.3, C | – | 84.5, C | – | 85.5, C | – |
| 3 | 71.8, CH | 4.25, dd (12.3, 5.3) | 71.7, CH | 4.46, dd (12.3, 5.0) | 66.9, CH | 4.49, dd (4.0, 2.0) | 68.6, CH | 4.74, dd (12.7, 5.1) |
| 4 | 23.7, CH2 | 2.15, m, Ha 2.08, m, Hb | 23.9, CH2 | 2.21, m, Ha 2.10, m, Hb | 22.9, CH2 | 2.18, m, Ha 2.00, m, Hb | 32.7, CH2 | 2.42, m, Ha 2.23, m, Hb |
| 5 | 27.5, CH2 | 2.65, m | 27.7, CH2 | 2.69, m | 24.4, CH2 | 2.84, m 2.44, m | 65.9, CH | 4.55, d (4.7) |
| 6 | 178.1, C | – | 178.5, C | – | 180.2, C | – | 174.0, C | – |
| 7 | 101.1, C | – | 101.6, C | – | 100.2, C | – | 101.8, C | – |
| 8 | 186.5, C | – | 186.7, C | – | 186.9, C | – | 188.0, C | – |
| 9 | 104.9, C | – | 105.4, C | – | 105.3, C | – | 105.8, C | – |
| 10 | 156.8, C | – | 160.1, C | – | 160.2, C | – | 160.1, C | – |
| 11 | 117.3, C | – | 114.2, CH | 6.13, s | 114.7, CH | 6.15, s | 114.3, CH | 6.14, s |
| 12 | 147.8, C | – | 147.6, C | – | 147.3, C | – | 148.5, C | – |
| 13 | 110.9, CH | 6.07, s | 115.3, C | – | 115.2, C | – | 115.5, C | – |
| 14 | 158.4, C | – | 154.8, C | – | 153.6, C | – | 155.0, C | – |
| 15 | 169.6, C | – | 169.8, C | – | 171.0, C | – | 169.2, C | – |
| 1′ | 72.8, CH | 5.96, s | 72.7, CH | 5.98, s | 72.7, CH | 5.98, s | 72.7, CH | 5.98, s |
| 2′ | 136.6, C | – | 136.5, C | – | 136.6, C | – | 136.5, C | – |
| 3′ | 123.1, CH | 6.89, s | 123.2, CH | 6.90, s | 123.2, CH | 6.90, s | 123.3, CH | 6.90, s |
| 4′ | 147.6, C | – | 147.7, C | – | 147.8, C | – | 147.8, C | – |
| 5′ | 119.3, CH | 6.81, s | 119.3, CH | 6.80, s | 119.3, CH | 6.80, s | 119.4, CH | 6.80, s |
| 6′ | 161.7, C | – | 161.6, C | – | 161.7, C | – | 161.5, C | – |
| 7′ | 112.7, C | – | 112.4, C | – | 112.6, C | – | 112.3, C | – |
| 8′ | 185.8, C | – | 185.3, C | – | 185.2, C | – | 184.8, C | – |
| 9′ | 105.5, C | – | 105.6, C | – | 105.7, C | – | 105.6, C | – |
| 10′ | 186.0, C | – | 186.1, C | – | 186.4, C | – | 186.4, C | – |
| 11′ | 37.3, CH | 4.76, dd (6.6, 1.0) | 38.6, CH | 4.81, dd (6.7, 0.7) | 37.9, CH | 4.76, dd (6.7, 1.0) | 38.7, CH | 4.83, dd (6.6, 0.9) |
| 12′ | 131.9, CH | 6.42, dd (8.5, 6.6) | 131.8, CH | 6.48, dd (8.4, 6.7) | 131.6, CH | 6.41, dd (8.4, 6.7) | 131.7, CH | 6.47, dd (8.4, 6.6) |
| 13′ | 132.4, CH | 6.05, dd (8.5, 1.0) | 132.3, CH | 6.08, dd (8.4, 0.7) | 132.7, CH | 6.07, dd (8.4, 1.0) | 132.4, CH | 6.08, d (8.4, 0.9) |
| 14′ | 41.5, C | – | 41.6, C | 41.4, C | – | 41.6, C | – | |
| 15′ | 35.1, CH2 | 2.74, d (18.5), Ha 2.68, d (18.5), Hb | 35.0, CH2 | 2.79, d (18.0), Ha 2.67, d (18.0), Hb | 35.0, CH2 | 2.78, d (17.9), Ha 2.68, d (17.9), Hb | 35.1, CH2 | 2.80, d (18.0), Ha 2.69, d (18.0), Hb |
| 16′ | 22.0, CH3 | 2.38, s | 22.1, CH3 | 2.38, s | 22.1, CH3 | 2.38, s | 22.1, CH3 | 2.38, s |
| 18′ | 173.1, C | – | 173.1, C | – | 173.0, C | – | 173.1, C | – |
| 19′ | 36.2, CH2 | 2.22, m | 36.2, CH2 | 2.23, m | 36.2, CH2 | 2.22, m | 36.2, CH2 | 2.22, m |
| 20′ | 18.4, CH2 | 1.57, m | 18.4, CH2 | 1.58, m | 18.4, CH2 | 1.57, m | 18.4, CH2 | 1.57, m |
| 21′ | 13.5, CH3 | 0.86, t (7.4) | 13.5, CH3 | 0.86, t (7.4) | 13.5, CH3 | 0.86, t (7.4) | 13.5, CH3 | 0.86, t (7.4) |
| 15- OCH3 | 53.4, CH3 | 3.67, s | 53.3, CH3 | 3.73, s | 53.6, CH3 | 3.74, s | 53.4, CH3 | 3.73, s |
| 3-OH | – | 14.14 | – | 14.26 | – | – | – | – |
| 6-OH | – | 13.84 | – | 13.95 | – | 14.10 | – | 13.70 |
| 10-OH | – | 11.76 | – | 11.05 | – | 11.24 | – | 10.91 |
| 6′-OH | – | 11.58 | – | 11.40 | – | 11.55 | – | 11.30 |
| 5 | 6 | 7 | ||||
|---|---|---|---|---|---|---|
| No | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) |
| 2 | 85.3, C | – | 84.9, C | – | 87.3, C | – |
| 3 | 71.8, CH | 4.46, dd (12.5, 5.0) | 80.8, CH | 5.01, dd (7.6, 6.8) | 73.8, CH | 4.23, dd (10.8, 1.8) |
| 4 | 23.9, CH2 | 2.23, m 2.13, m | 22.2, CH2 | 2.42, m | 25.6, CH2 | 1.97, m 1.78, m |
| 5 | 27.7, CH2 | 2.70, m | 27.7, CH2 | 2.67, m | 30.0, CH2 | 2.69, m |
| 6 | 178.5, C | – | 175.2, C | – | 177.2, C | – |
| 7 | 101.6, C | – | 38.6, CH2 | 3.21, d (17.0) 3.04, d (17.0) | 38.2, CH2 | 3.20, s |
| 8 | 186.8, C | – | 194.1, C | – | 195.7, C | – |
| 9 | 105.4, C | – | 105.8, C | – | 105.9, C | – |
| 10 | 160.1, C | – | 160.1, C | – | 160.1, C | – |
| 11 | 114.3, CH | 6.14, s | 114.9, CH | 6.16, s | 114.1, CH | 6.14, s |
| 12 | 147.6, C | – | 148.9, C | – | 147.8, C | – |
| 13 | 115.3, C | – | 114.9, C | – | 114.8, C | – |
| 14 | 154.7, C | – | 154.8, C | – | 155.1, C | – |
| 15 | 169.8, C | – | 168.7, C | – | 170.0, C | – |
| 1′ | 73.0, CH | 5.97, s | 72.6, CH | 5.98, s | 72.6, CH | 5.98, s |
| 2′ | 136.4, C | – | 136.5, C | – | 136.5, C | – |
| 3′ | 123.3, CH | 6.90, s | 123.2, CH | 6.89, s | 123.2, CH | 6.90, s |
| 4′ | 147.8, C | – | 147.8, C | – | 147.8, C | – |
| 5′ | 119.4, CH | 6.81, s | 119.3, CH | 6.80, s | 119.3, CH | 6.80, s |
| 6′ | 161.7, C | – | 161.8, C | – | 161.7, C | – |
| 7′ | 112.5, C | – | 112.7, C | – | 112.6, C | – |
| 8′ | 185.6, C | – | 184.8, C | – | 185.4, C | – |
| 9′ | 105.6, C | – | 105.4, C | – | 105.5, C | – |
| 10′ | 185.9, C | – | 186.6, C | – | 186.2, C | – |
| 11′ | 38.6, CH | 4.79, d (6.7) | 37.9, CH | 4.65, dd (6.6, 0.9) | 38.0, CH | 4.73, dd (6.6, 0.6) |
| 12′ | 131.8, CH | 6.49, dd (8.5, 6.7) | 131.4, CH | 6.41, dd (8.5, 6.6) | 131.4, CH | 6.42, dd (8.5, 6.6) |
| 13′ | 132.2, CH | 6.09, d (8.5) | 132.9, CH | 6.09, d (8.5, 0.9) | 132.9, CH | 6.09, dd (8.5, 0.6) |
| 14′ | 41.5, C | – | 41.5, C | – | 41.4, C | – |
| 15′ | 35.0, CH2 | 2.79, d (17.5) 2.68, d (17.5) | 35.1, CH2 | 2.79, d (18.2) 2.68, d (18.2) | 35.1, CH2 | 2.79, d (17.5) 2.67, d (17.5) |
| 16′ | 22.1, CH3 | 2.38, s | 22.1, CH3 | 2.38, s | 22.1, CH3 | 2.38, s |
| 18′ | 170.4, C | – | 173.1, C | – | 173.1, C | – |
| 19′ | 21.1, CH3 | 2.02, s | 36.2, CH2 | 2.22, m | 36.2, CH2 | 2.21, m |
| 20′ | – | – | 18.4, CH2 | 1.57, m | 18.4, CH2 | 1.58, m |
| 21′ | – | – | 13.5, CH3 | 0.86, t (7.4) | 13. 5, CH3 | 0.86, t (7.4) |
| 15- OCH3 | 53.3, CH3 | 3.74, s | 53.8, CH3 | 3.75, s | 53.4, CH3 | 3.73, s |
| 3-OH | – | – | – | – | – | – |
| 6-OH | – | 13.95 | – | – | – | – |
| 10-OH | – | 11.05 | – | 11.36 | – | 11.5 |
| 6′-OH | – | 11.45 | – | 11.60 | – | – |
| Test Organism | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Positive Control |
|---|---|---|---|---|---|---|---|---|---|
| Schizosaccharomyces pombe | – | – | 66.70 | – | – | – | – | – | 33.30 1 |
| Pichia anomala | – | – | 8.30 | – | – | – | – | – | 33.30 1 |
| Mucor hiemalis | 66.70 | – | 2.10 | 66.70 | 66.70 | 66.70 | 66.70 | – | 33.30 1 |
| Candida albicans | – | – | 16.70 | – | – | – | – | – | 33.30 1 |
| Rhodotorula glutinis | – | – | 2.10 | 66.70 | – | – | – | – | 16.70 1 |
| Micrococcus luteus | 4.20 | 2.10 | 2.10 | 2.10 | 2.10 | 8.30 | 4.20 | 8.30 | 0.80 2 |
| Bacillus subtilis | 0.40 | 0.40 | 0.20 | 0.80 | 0.20 | 4.20 | 4.20 | 2.10 | 8.30 2 |
| Staphylococcus aureus | 2.10 | 2.10 | 1.00 | 2.10 | 8.30 | 4.20 | 8.30 | 8.30 | 0.40 2 |
| Mycobacterium smegmatis | – | – | – | – | – | – | – | – | 1.70 3 |
| Escherichia coli | – | – | – | – | – | – | – | – | 3.30 2 |
| Pseudomonas aeruginosa | – | – | – | – | – | – | – | – | 0.40 4 |
| Chromobacterium violaceum | – | – | – | – | – | – | – | 66.70 | 0.80 2 |
| Compound | KB 3.1 | L929 | A549 | SK-OV-3 | PC-3 | A431 | MCF-7 |
|---|---|---|---|---|---|---|---|
| 1 | 0.20 | 1.13 | 0.28 | 0.14 | 0.71 | 0.05 | 0.06 |
| 2 | 0.19 | 1.46 | 0.40 | 0.13 | 1.06 | 0.06 | 0.04 |
| 3 | 0.15 | 0.98 | 0.26 | 0.15 | 0.65 | 0.05 | 0.04 |
| 4 | 5.76 | 13.48 | 9.39 | 4.09 | 8.18 | 2.27 | 1.97 |
| 5 | 0.19 | 1.12 | 0.29 | 0.14 | 0.62 | 0.06 | 0.03 |
| 6 | 3.42 | 10.56 | 18.63 | 3.88 | 5.43 | 3.11 | 1.10 |
| 7 | 3.47 | 9.97 | 11.48 | 3.93 | 4.98 | 2.87 | 1.03 |
| 8 | 1.06 | 4.70 | 1.22 | 0.47 | 3.14 | 0.16 | 0.10 |
| epothilon B | 0.00003 | 0.00051 | 0.00009 | 0.00009 | 0.00007 | 0.00005 | 0.00003 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, L.; Marin-Felix, Y.; Surup, F.; Stchigel, A.M.; Stadler, M. Seven New Cytotoxic and Antimicrobial Xanthoquinodins from Jugulospora vestita. J. Fungi 2020, 6, 188. https://doi.org/10.3390/jof6040188
Shao L, Marin-Felix Y, Surup F, Stchigel AM, Stadler M. Seven New Cytotoxic and Antimicrobial Xanthoquinodins from Jugulospora vestita. Journal of Fungi. 2020; 6(4):188. https://doi.org/10.3390/jof6040188
Chicago/Turabian StyleShao, Lulu, Yasmina Marin-Felix, Frank Surup, Alberto M. Stchigel, and Marc Stadler. 2020. "Seven New Cytotoxic and Antimicrobial Xanthoquinodins from Jugulospora vestita" Journal of Fungi 6, no. 4: 188. https://doi.org/10.3390/jof6040188
APA StyleShao, L., Marin-Felix, Y., Surup, F., Stchigel, A. M., & Stadler, M. (2020). Seven New Cytotoxic and Antimicrobial Xanthoquinodins from Jugulospora vestita. Journal of Fungi, 6(4), 188. https://doi.org/10.3390/jof6040188