
The Response of Airborne Mycobiome to Dust Storms in the Eastern Mediterranean


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Sequencing
2.3. Analysis of Metagenomes
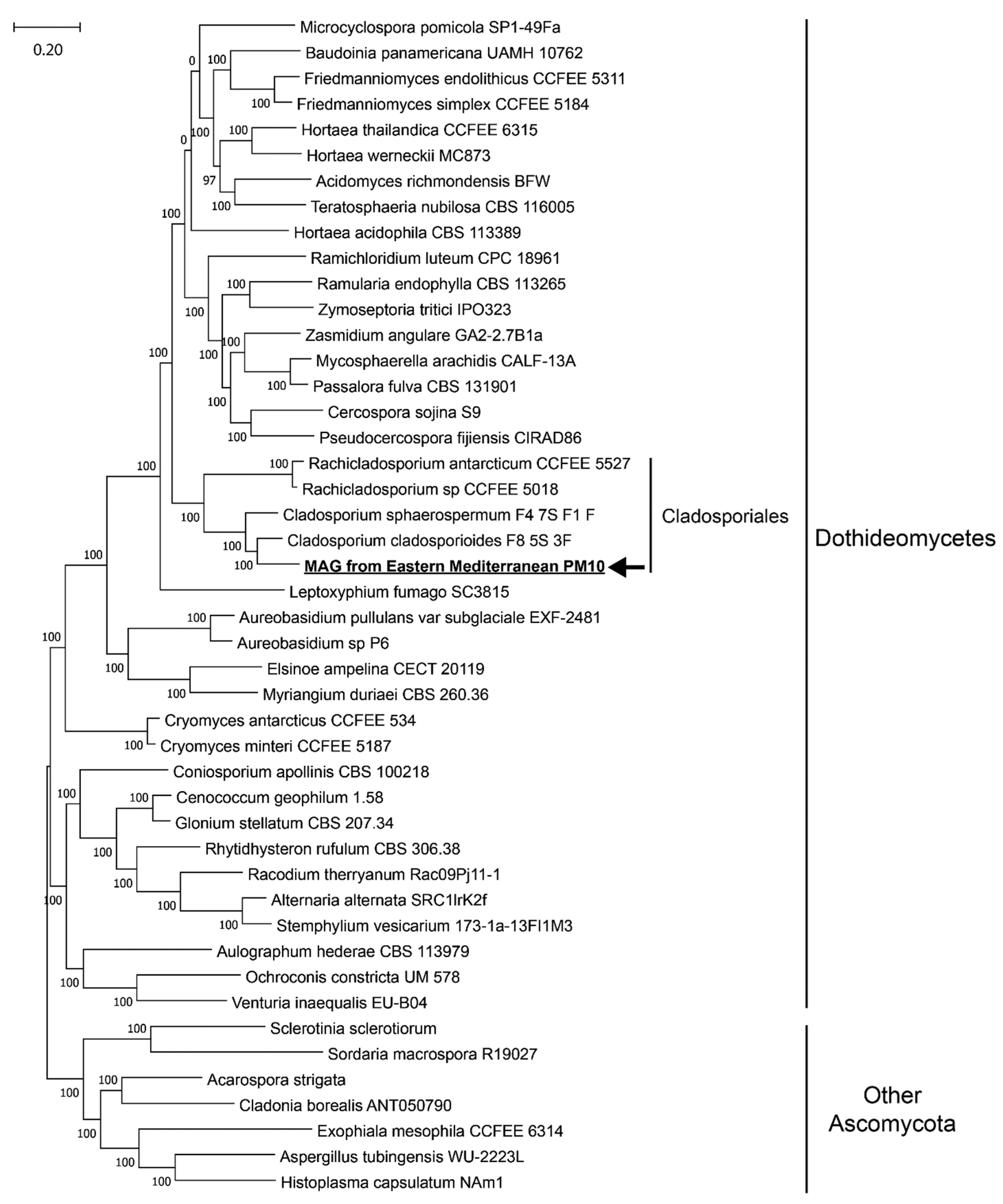
2.4. Fungal Metagenome-Assembled Genome
2.5. Statistical Analysis
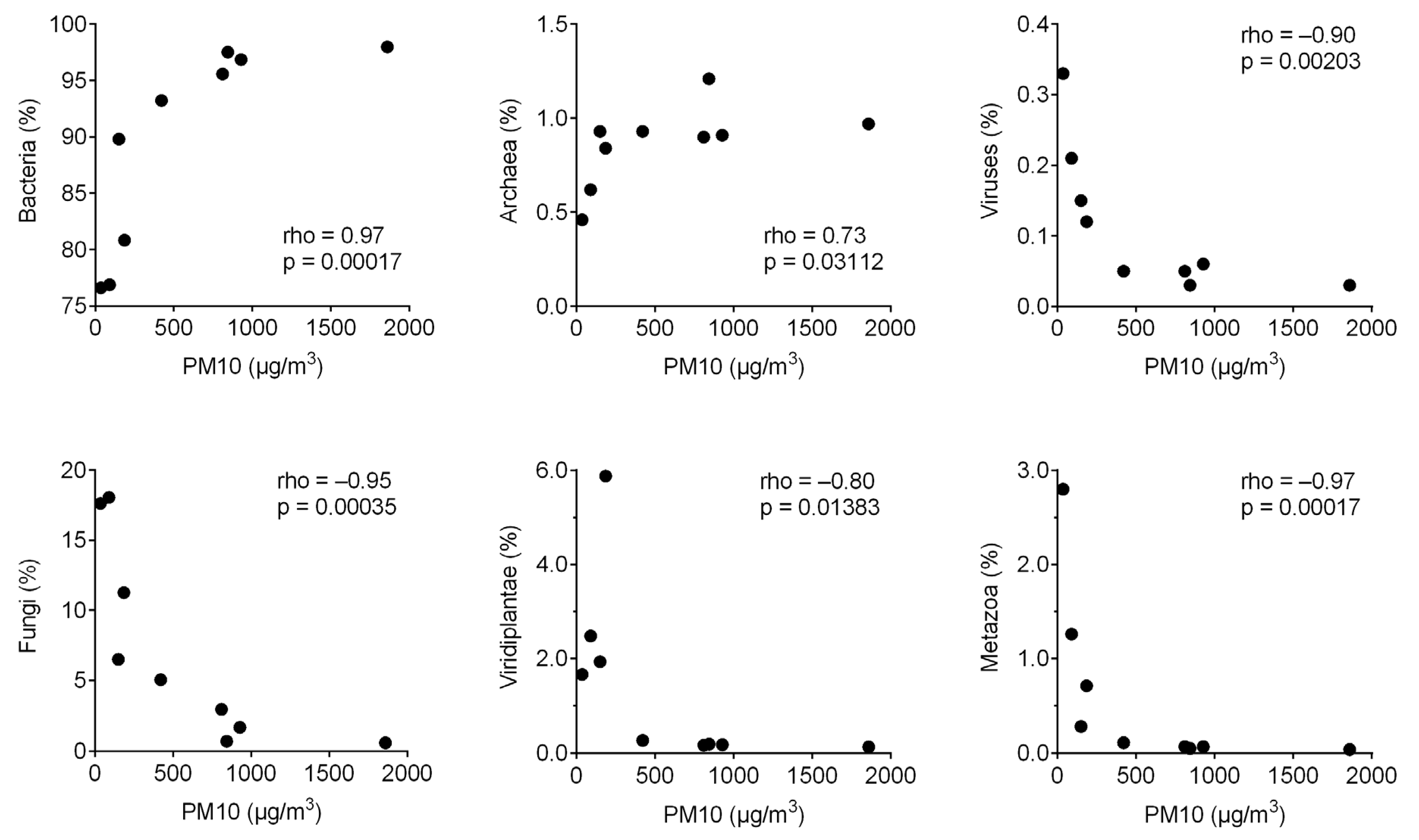
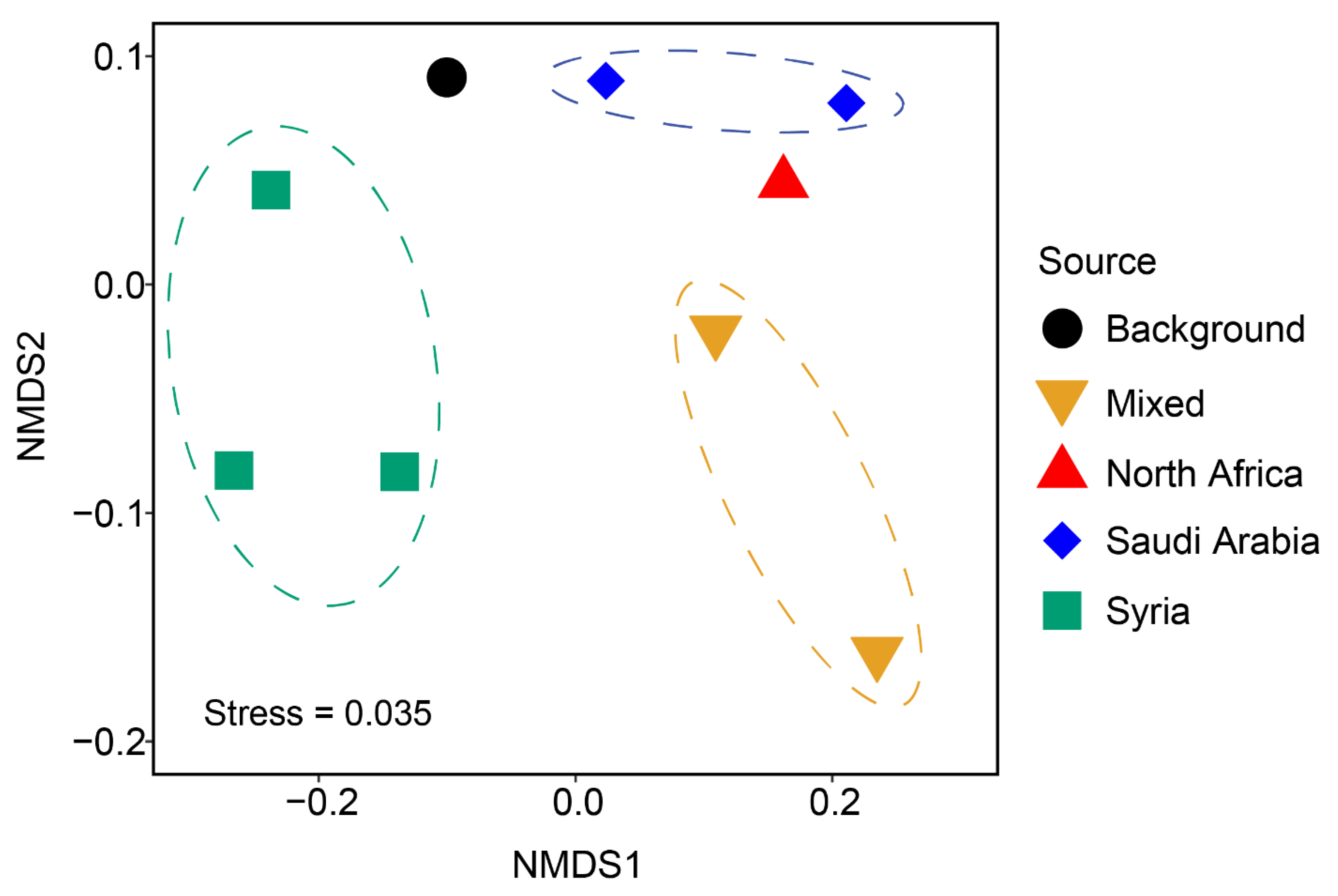
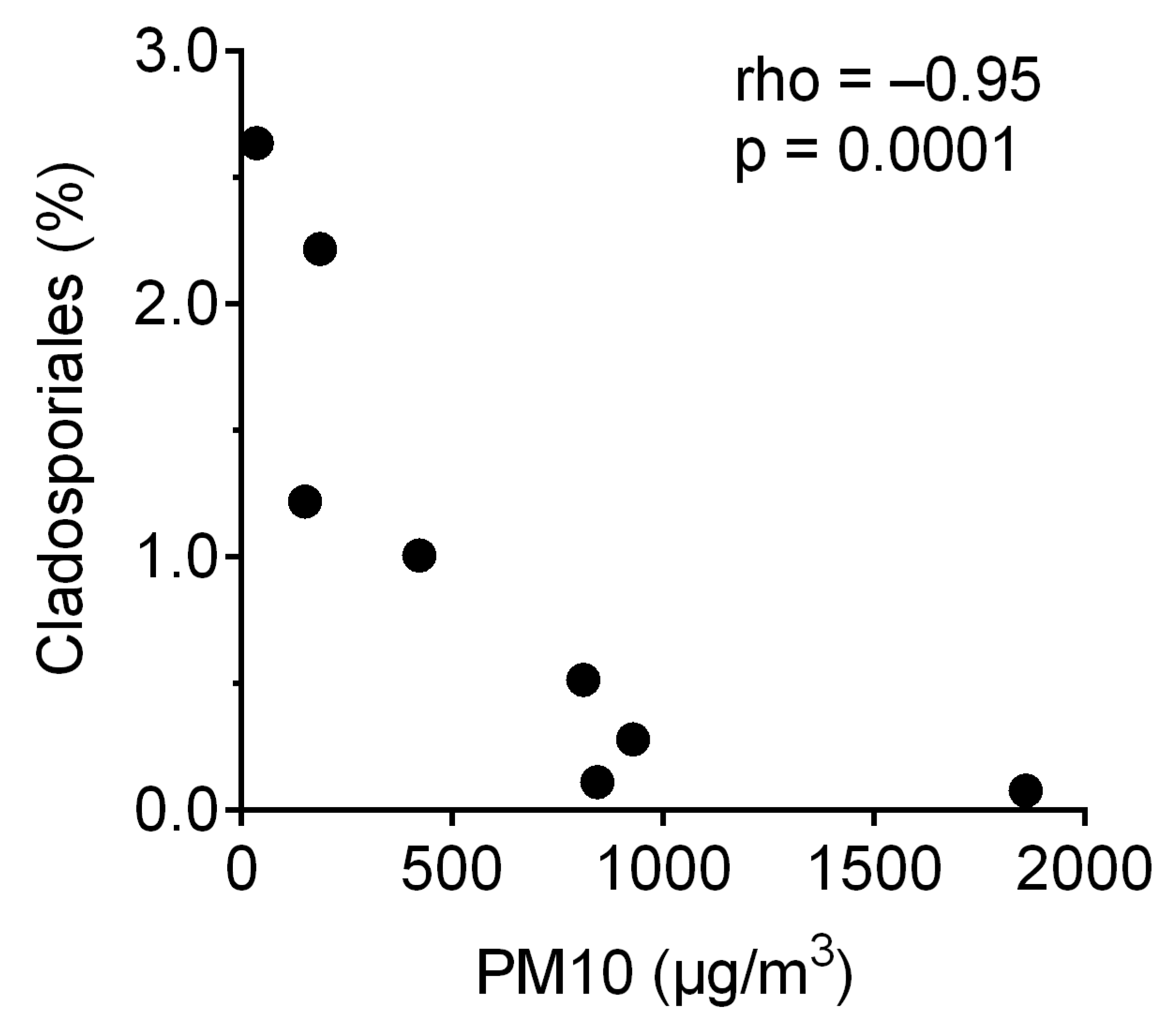
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jaenicke, R. Abundance of Cellular Material and Proteins in the Atmosphere. Science 2005, 308, 73. [Google Scholar] [CrossRef]
- Rahav, E.; Paytan, A.; Mescioglu, E.; Bar-Zeev, E.; Martínez Ruiz, F.; Xian, P.; Herut, B. Bio-Aerosols Negatively Affect Prochlorococcus in Oligotrophic Aerosol-Rich Marine Regions. Atmosphere 2020, 11, 540. [Google Scholar] [CrossRef]
- Mescioglu, E.; Rahav, E.; Frada, M.J.; Rosenfeld, S.; Raveh, O.; Galletti, Y.; Santinelli, C.; Herut, B.; Paytan, A. Dust-Associated Airborne Microbes Affect Primary and Bacterial Production Rates, and Eukaryotes Diversity, in the Northern Red Sea: A Mesocosm Approach. Atmosphere 2019, 10, 358. [Google Scholar] [CrossRef] [Green Version]
- Rahav, E.; Belkin, N.; Paytan, A.; Herut, B. Phytoplankton and Bacterial Response to Desert Dust Deposition in the Coastal Waters of the Southeastern Mediterranean Sea: A Four-Year In Situ Survey. Atmosphere 2018, 9, 305. [Google Scholar] [CrossRef] [Green Version]
- Griffin, D.W.; Kellogg, C.A.; Shinn, E.A. Dust in the Wind: Long Range Transport of Dust in the Atmosphere and Its Implications for Global Public and Ecosystem Health. Glob. Chang. Hum. Health 2001, 2, 20–33. [Google Scholar] [CrossRef]
- Zhai, Y.; Li, X.; Wang, T.; Wang, B.; Li, C.; Zeng, G. A review on airborne microorganisms in particulate matters: Composition, characteristics and influence factors. Environ. Int. 2018, 113, 74–90. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.C.; Christner, B.C.; Guzik, T.G.; Granger, D.J.; Stewart, M.F. Abundance and survival of microbial aerosols in the troposphere and stratosphere. ISME J. 2019, 13, 2789–2799. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Lee, K.C.; Kawai, K.; Onishi, K.; Hong, C.S.; Kurosaki, Y.; Shinoda, M.; Kai, K.; Iwasaka, Y.; Archer, S.D.J.; et al. Aeolian Dispersal of Bacteria Associated With Desert Dust and Anthropogenic Particles Over Continental and Oceanic Surfaces. J. Geophys. Res. Atmos. 2019, 124, 5579–5588. [Google Scholar] [CrossRef]
- Spracklen, D.V.; Heald, C.L. The contribution of fungal spores and bacteria to regional and global aerosol number and ice nucleation immersion freezing rates. Atmos. Chem. Phys. 2014, 14, 9051–9059. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.J.; Coe, A.; Dove, L.A.; Zawadowicz, M.A.; Dooley, K.; Biller, S.J.; Zhang, Y.; Chisholm, S.W.; Cziczo, D.J. Investigating the Heterogeneous Ice Nucleation of Sea Spray Aerosols Using Prochlorococcus as a Model Source of Marine Organic Matter. Environ. Sci. Technol. 2019, 53, 1139–1149. [Google Scholar] [CrossRef]
- Gandolfi, I.; Bertolini, V.; Ambrosini, R.; Bestetti, G.; Franzetti, A. Unravelling the bacterial diversity in the atmosphere. Appl. Microbiol. Biotechnol. 2013, 97, 4727–4736. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich-Nowoisky, J.; Ruzene Nespoli, C.; Pickersgill, D.A.; Galand, P.E.; Müller-Germann, I.; Nunes, T.; Gomes Cardoso, J.; Almeida, S.M.; Pio, C.; Andreae, M.O.; et al. Diversity and seasonal dynamics of airborne archaea. Biogeosciences 2014, 11, 6067–6079. [Google Scholar] [CrossRef] [Green Version]
- Barberán, A.; Ladau, J.; Leff, J.W.; Pollard, K.S.; Menninger, H.L.; Dunn, R.R.; Fierer, N. Continental-scale distributions of dust-associated bacteria and fungi. Proc. Natl. Acad. Sci. USA 2015, 112, 5756–5761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayol, E.; Arrieta, J.M.; Jiménez, M.A.; Martínez-Asensio, A.; Garcias-Bonet, N.; Dachs, J.; González-Gaya, B.; Royer, S.-J.; Benítez-Barrios, V.M.; Fraile-Nuez, E.; et al. Long-range transport of airborne microbes over the global tropical and subtropical ocean. Nat. Commun. 2017, 8, 201. [Google Scholar] [CrossRef] [PubMed]
- Mescioglu, E.; Rahav, E.; Belkin, N.; Xian, P.; Eizenga, J.M.; Vichik, A.; Herut, B.; Paytan, A. Aerosol Microbiome over the Mediterranean Sea Diversity and Abundance. Atmosphere 2019, 10, 440. [Google Scholar] [CrossRef] [Green Version]
- Behzad, H.; Gojobori, T.; Mineta, K. Challenges and Opportunities of Airborne Metagenomics. Genome Biol. Evol. 2015, 7, 1216–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.; Xu, C.; Xu, X.; Zhu, C.; Li, J.; Lv, G. Characteristics of atmospheric bacterial and fungal communities in PM2.5 following biomass burning disturbance in a rural area of North China Plain. Sci. Total Environ. 2019, 651, 2727–2739. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.; Lee, T.K.; Choi, E.J.; Yang, J.; Shukla, S.K.; Hwang, S.-I.; Park, J. Molecular approaches for the detection and monitoring of microbial communities in bioaerosols: A review. J. Environ. Sci. 2017, 51, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Be, N.A.; Thissen, J.B.; Fofanov, V.Y.; Allen, J.E.; Rojas, M.; Golovko, G.; Fofanov, Y.; Koshinsky, H.; Jaing, C.J. Metagenomic Analysis of the Airborne Environment in Urban Spaces. Microb. Ecol. 2015, 69, 346–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, P.T.; Probst, A.J.; Grigoriev, I.V.; Thomas, B.C.; Banfield, J.F. Genome-reconstruction for eukaryotes from complex natural microbial communities. Genome Res. 2018, 28, 569–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.; Wilken, S.E.; Lankiewicz, T.S.; Gilmore, S.P.; Brown, J.L.; Henske, J.K.; Swift, C.L.; Salamov, A.; Barry, K.; Grigoriev, I.V.; et al. Genomic and functional analyses of fungal and bacterial consortia that enable lignocellulose breakdown in goat gut microbiomes. Nat. Microbiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Goudie, A.S. Dust storms: Recent developments. J. Environ. Manag. 2009, 90, 89–94. [Google Scholar] [CrossRef]
- Achilleos, S.; Mouzourides, P.; Kalivitis, N.; Katra, I.; Kloog, I.; Kouis, P.; Middleton, N.; Mihalopoulos, N.; Neophytou, M.; Panayiotou, A.; et al. Spatio-temporal variability of desert dust storms in Eastern Mediterranean (Crete, Cyprus, Israel) between 2006 and 2017 using a uniform methodology. Sci. Total Environ. 2020, 714, 136693. [Google Scholar] [CrossRef] [PubMed]
- Gat, D.; Mazar, Y.; Cytryn, E.; Rudich, Y. Origin-Dependent Variations in the Atmospheric Microbiome Community in Eastern Mediterranean Dust Storms. Environ. Sci. Technol. 2017, 51, 6709–6718. [Google Scholar] [CrossRef]
- Lang-Yona, N.; Öztürk, F.; Gat, D.; Aktürk, M.; Dikmen, E.; Zarmpas, P.; Tsagkaraki, M.; Mihalopoulos, N.; Birgül, A.; Kurt-Karakuş, P.B.; et al. Links between airborne microbiome, meteorology, and chemical composition in northwestern Turkey. Sci. Total Environ. 2020, 725, 138227. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-Y.; Fong, J.J.; Park, M.S.; Chang, L.; Lim, Y.W. Identifying airborne fungi in Seoul, Korea using metagenomics. J. Microbiol. 2014, 52, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Acker, J.G.; Leptoukh, G. Online analysis enhances use of NASA Earth science data. Eos Trans. Am. Geophys. Union 2007, 88, 14–17. [Google Scholar] [CrossRef]
- Krasnov, H.; Katra, I.; Friger, M. Increase in dust storm related PM10 concentrations: A time series analysis of 2001–2015. Environ. Pollut. 2016, 213, 36–42. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap Short-Read Aligner, and other Bioinformatics Tools, 2015.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Spatafora, J.W.; Aime, M.C.; Grigoriev, I.V.; Martin, F.; Stajich, J.E.; Blackwell, M. The Fungal Tree of Life: From Molecular Systematics to Genome-Scale Phylogenies. In The Fungal Kingdom; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 1–34. [Google Scholar]
- Tedersoo, L.; Sánchez-Ramírez, S.; Kõljalg, U.; Bahram, M.; Döring, M.; Schigel, D.; May, T.; Ryberg, M.; Abarenkov, K. High-level classification of the Fungi and a tool for evolutionary ecological analyses. Fungal Divers. 2018, 90, 135–159. [Google Scholar] [CrossRef] [Green Version]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.; Otillar, R.; Riley, R.; Salamov, A.; Zhao, X.; Korzeniewski, F.; et al. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014, 42, D699–D704. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef]
- Kang, D.; Li, F.; Kirton, E.S.; Thomas, A.; Egan, R.S.; An, H.; Wang, Z. MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Zdobnov, E.M.; Tegenfeldt, F.; Kuznetsov, D.; Waterhouse, R.M.; Simão, F.A.; Ioannidis, P.; Seppey, M.; Loetscher, A.; Kriventseva, E.V. OrthoDB v9.1: Cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 2017, 45, D744–D749. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.M. Funannotate: A Fungal Genome Annotation and Comparative Genomics Pipeline, 2016.
- Frith, M.C. A new repeat-masking method enables specific detection of homologous sequences. Nucleic Acids Res. 2011, 39, e23. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef] [Green Version]
- Abdollahzadeh, J.; Groenewald, J.Z.; Coetzee, M.P.A.; Wingfield, M.J.; Crous, P.W. Evolution of lifestyles in Capnodiales. Stud. Mycol. 2020, 95, 381–414. [Google Scholar] [CrossRef]
- Peng, X.; Valentine, D.L. Diversity and N2O Production Potential of Fungi in an Oceanic Oxygen Minimum Zone. J. Fungi 2021, 7, 218. [Google Scholar] [CrossRef]
- Li, Y.; Fu, H.; Wang, W.; Liu, J.; Meng, Q.; Wang, W. Characteristics of bacterial and fungal aerosols during the autumn haze days in Xi’an, China. Atmos. Environ. 2015, 122, 439–447. [Google Scholar] [CrossRef]
- DeLeon-Rodriguez, N.; Lathem, T.L.; Rodriguez-R, L.M.; Barazesh, J.M.; Anderson, B.E.; Beyersdorf, A.J.; Ziemba, L.D.; Bergin, M.; Nenes, A.; Konstantinidis, K.T. Microbiome of the upper troposphere: Species composition and prevalence, effects of tropical storms, and atmospheric implications. Proc. Natl. Acad. Sci. USA 2013, 110, 2575–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, R.; Begerow, D.; Oberwinkler, E.; Piepenbring, M.; Berbee, M.L. Ustilaginomycetes. In Systematics and Evolution; McLaughlin, D.J., McLaughlin, E.G., Lemke, P.A., Eds.; The Mycota; Springer: Berlin/Heidelberg, Germany, 2001; pp. 57–83. [Google Scholar]
- Padamsee, M.; Kumar, T.K.A.; Riley, R.; Binder, M.; Boyd, A.; Calvo, A.M.; Furukawa, K.; Hesse, C.; Hohmann, S.; James, T.Y.; et al. The genome of the xerotolerant mold Wallemia sebi reveals adaptations to osmotic stress and suggests cryptic sexual reproduction. Fungal Genet. Biol. 2012, 49, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelton, B.G.; Kirkland, K.H.; Flanders, W.D.; Morris, G.K. Profiles of Airborne Fungi in Buildings and Outdoor Environments in the United States. Appl. Environ. Microbiol. 2002, 68, 1743–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensch, K.; Groenewald, J.Z.; Braun, U.; Dijksterhuis, J.; de Jesús Yáñez-Morales, M.; Crous, P.W. Common but different: The expanding realm of Cladosporium. Stud. Mycol. 2015, 82, 23–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleine, C.; Masonjones, S.; Selbmann, L.; Zucconi, L.; Onofri, S.; Pacelli, C.; Stajich, J.E. Draft Genome Sequences of the Antarctic Endolithic Fungi Rachicladosporium antarcticum CCFEE 5527 and Rachicladosporium sp. CCFEE 5018. Genome Announc. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensch, K.; Groenewald, J.Z.; Meijer, M.; Dijksterhuis, J.; Jurjević, Ž.; Andersen, B.; Houbraken, J.; Crous, P.W.; Samson, R.A. Cladosporium species in indoor environments. Stud. Mycol. 2018, 89, 177–301. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Average% | Rho | p-Value |
|---|---|---|---|
| Dothideomycetes | 49.91% | −0.65 | 0.06656 |
| Eurotiomycetes | 16.78% | 0.216667 | 0.5809 |
| Sordariomycetes | 8.55% | 0.15 | 0.7081 |
| Leotiomycetes | 2.01% | −0.36667 | 0.3363 |
| Saccharomycetes | 1.23% | 0.733333 | 0.03112 |
| Pezizomycetes | 0.40% | 0.383333 | 0.3125 |
| Lecanoromycetes | 0.31% | 0.333333 | 0.3853 |
| Orbiliomycetes | 0.15% | 0.233333 | 0.5517 |
| Taphrinomycetes | 0.11% | −0.11667 | 0.7756 |
| Xylonomycetes | 0.10% | −0.11667 | 0.7756 |
| Agaricomycetes | 5.78% | 0.75 | 0.02549 |
| Ustilaginomycetes | 4.72% | 0.15 | 0.7081 |
| Wallemiomycetes | 4.66% | 0.5 | 0.1777 |
| Tremellomycetes | 2.12% | 0.333333 | 0.3853 |
| Exobasidiomycetes | 0.56% | −0.23333 | 0.5517 |
| Microbotryomycetes | 0.25% | −0.61667 | 0.08573 |
| Pucciniomycetes | 0.14% | 0.183333 | 0.6436 |
| Microsporidia | 0.69% | 0.833333 | 0.008267 |
| Mucoromycotina | 0.62% | 0.216667 | 0.5809 |
| Chytridiomycota | 0.18% | 0.766667 | 0.02139 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, X.; Gat, D.; Paytan, A.; Rudich, Y. The Response of Airborne Mycobiome to Dust Storms in the Eastern Mediterranean. J. Fungi 2021, 7, 802. https://doi.org/10.3390/jof7100802
Peng X, Gat D, Paytan A, Rudich Y. The Response of Airborne Mycobiome to Dust Storms in the Eastern Mediterranean. Journal of Fungi. 2021; 7(10):802. https://doi.org/10.3390/jof7100802
Chicago/Turabian StylePeng, Xuefeng, Daniela Gat, Adina Paytan, and Yinon Rudich. 2021. "The Response of Airborne Mycobiome to Dust Storms in the Eastern Mediterranean" Journal of Fungi 7, no. 10: 802. https://doi.org/10.3390/jof7100802
APA StylePeng, X., Gat, D., Paytan, A., & Rudich, Y. (2021). The Response of Airborne Mycobiome to Dust Storms in the Eastern Mediterranean. Journal of Fungi, 7(10), 802. https://doi.org/10.3390/jof7100802