
Comparative Proteomic Analysis within the Developmental Stages of the Mushroom White Hypsizygus marmoreus



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture Conditions and Acquisition of the White H. marmoreus Samples
2.2. Protein Extraction and Peptide Digestion
2.3. Label-Free LC-MS/MS Quantitative Proteomics Analysis
2.4. Peptides and Proteins Identification
2.5. Bioinformation Analysis
3. Results
3.1. Label-Free LC-MS/MS Quantitative Proteomics Analysis for the Six Groups of the White H. marmoreus Samples and the Correlation among the Six Groups of Samples
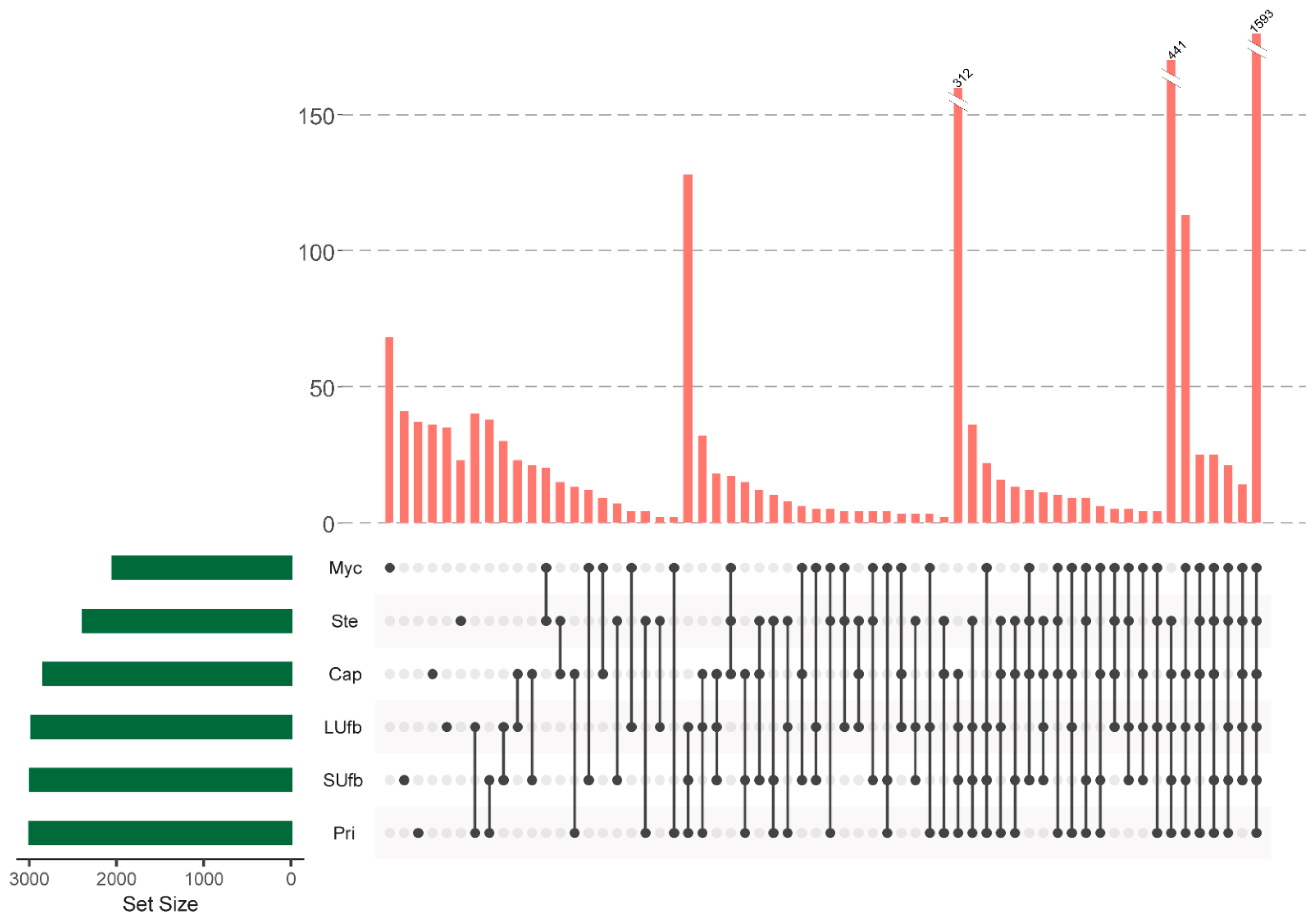
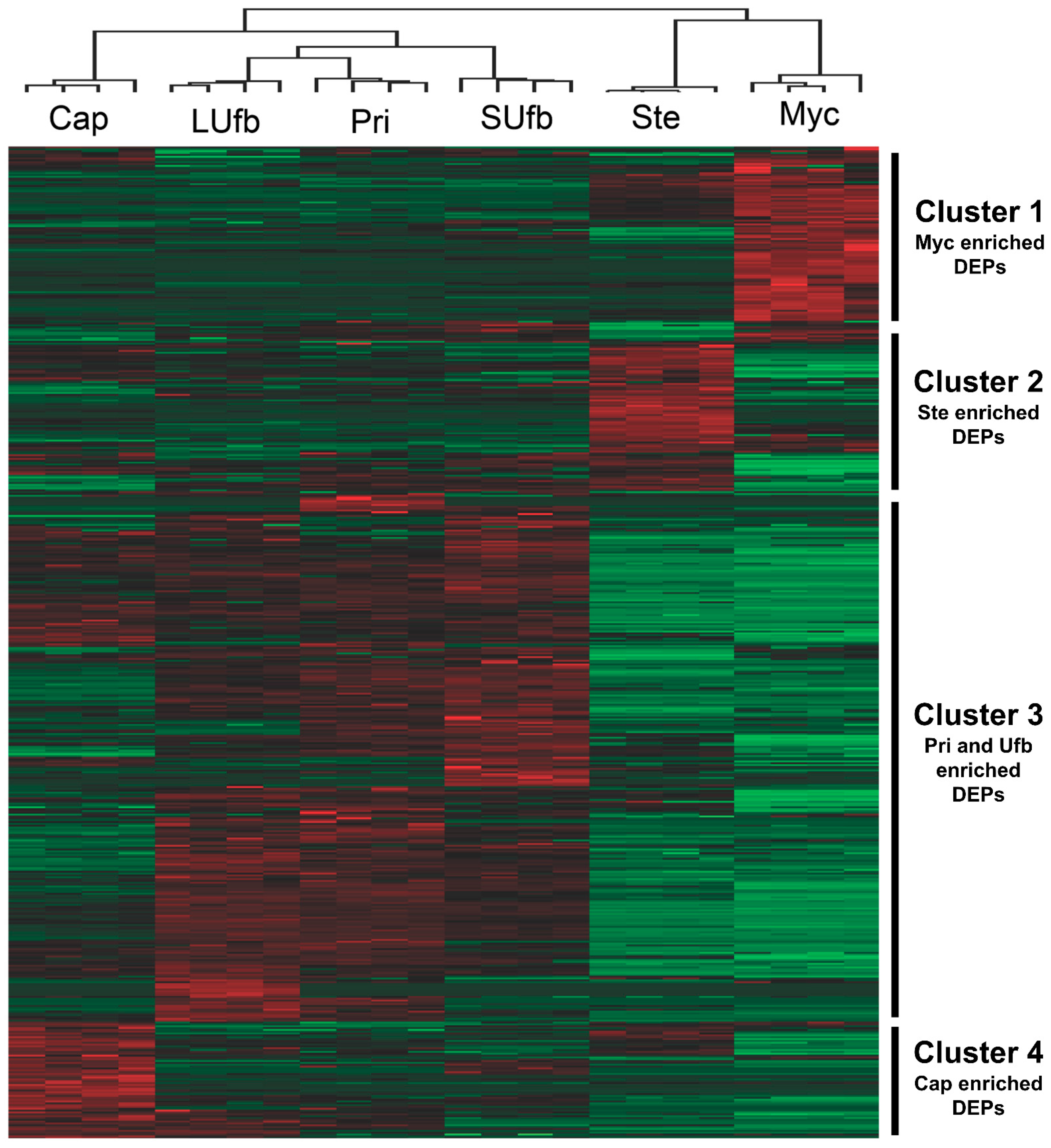
3.2. Six Groups of Samples Intersection and Quantitative Visualization of DEPs
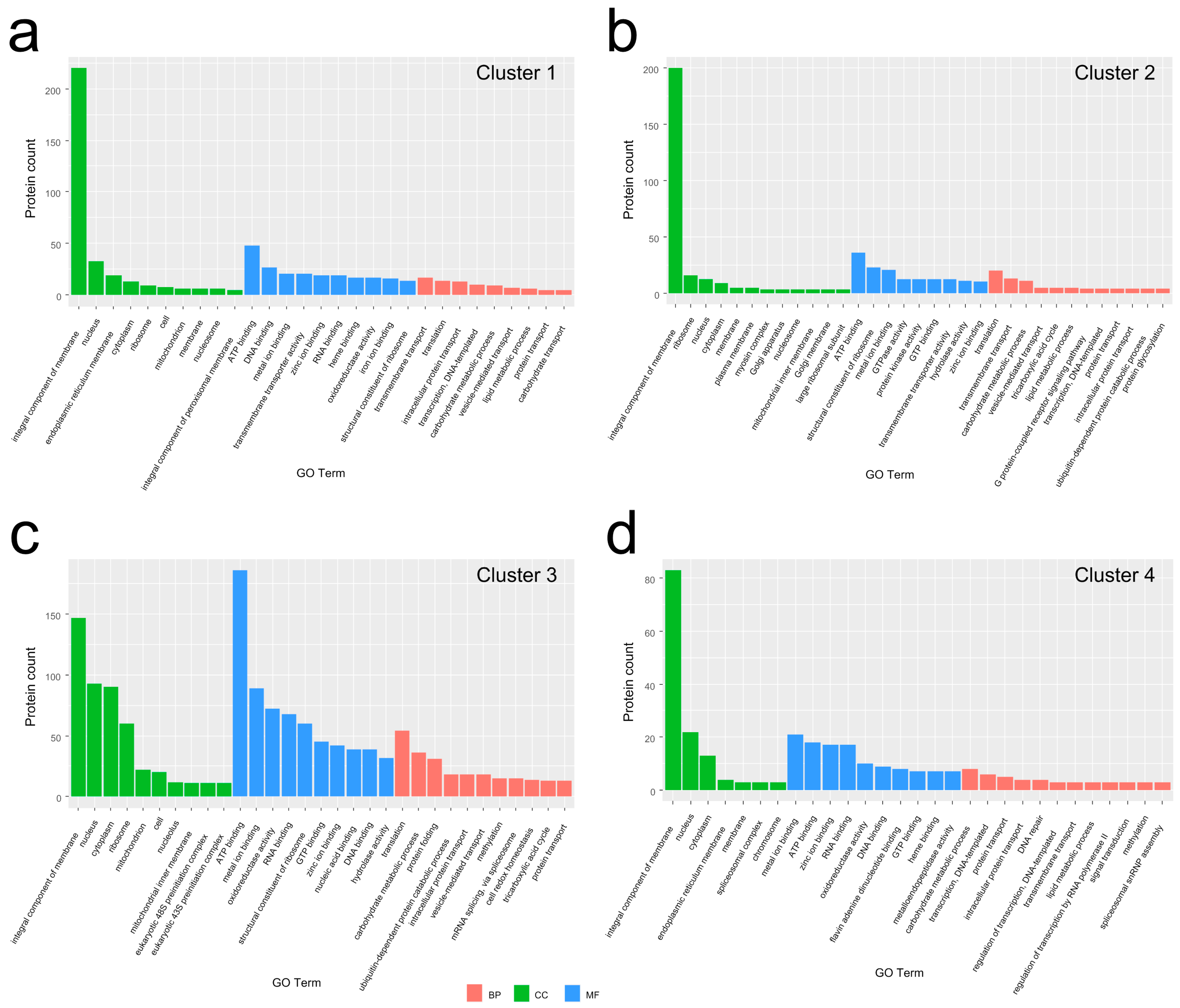
3.3. GO Analysis for the DEPs in Each Group to Molecular Function (MF), Cellular Component (CC), and Biological Process (BP) Categories
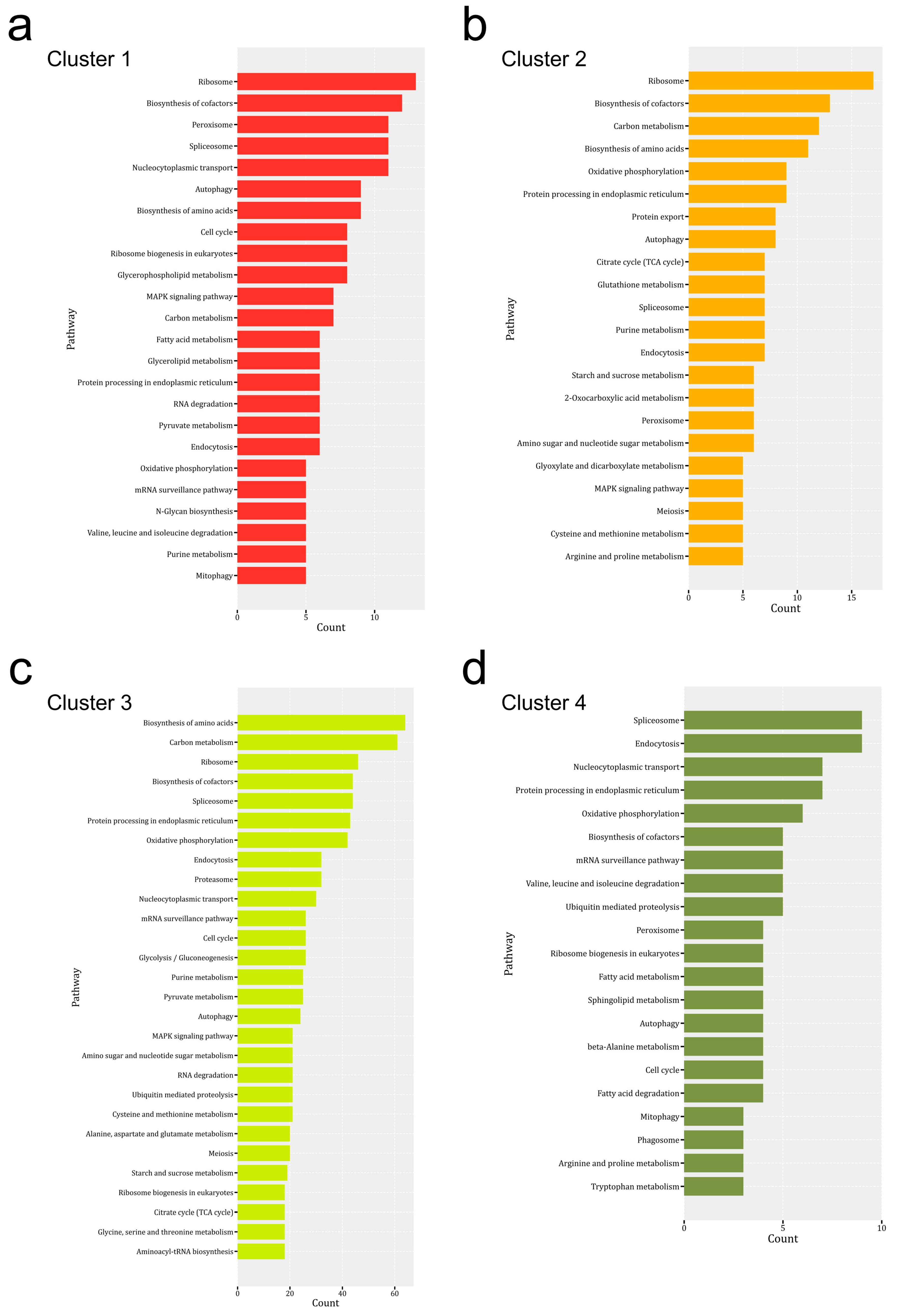
3.4. KEGG Pathway Enrichment Analysis of DEPs from Mycelium to the Mature Fruiting Body
4. Discussion
4.1. The Proteome and Transcriptome of H. marmoreus Exhibit the Same Major Enriched Pathways and the Enriched Pathways Are Similar among Some Different Mushrooms
4.2. MAPK and cAMP Signaling Pathways Play Vital Roles in the Growth and Development Process
4.3. Identified CAZyme Families and Their Functions in the Growth and Development Process
4.4. Important TFs and Their Functions in the Growth and Development Process
4.5. HSPs and Their Functions in the Growth and Development Process
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, G.; Lin, J.; Shi, Y.; Chang, X.; Wang, Y.; Guo, L.; Wang, W.; Dou, M.; Deng, Y.; Ming, R. Mitochondrial genome in Hypsizygus marmoreus and its evolution in Dikarya. BMC Genom. 2019, 20, 765. [Google Scholar] [CrossRef]
- Son, S.Y.; Park, Y.J.; Jung, E.S.; Singh, D.; Lee, Y.W.; Kim, J.G.; Lee, C.H. Integrated metabolomics and transcriptomics unravel the metabolic pathway variations for different sized beech mushrooms. Int. J. Mol. Sci. 2019, 20, 6007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mleczek, M.; Siwulski, M.; Rzymski, P.; Budka, A.; Kala, P.; Jasińska, A.; Gsecka, M.; Budzyńska, S.; Niedzielski, P. Comparison of elemental composition of mushroom Hypsizygus marmoreus originating from commercial production and experimental cultivation. Sci. Hortic. 2018, 236, 30–35. [Google Scholar] [CrossRef]
- Xu, Q.; Wang, H.; Li, T.; Chen, L.; Liu, R. Comparison of phenolics, antioxidant, and antiproliferative activities of two Hypsizygus marmoreus varieties. J. Food Sci. 2020, 85, 2227–2235. [Google Scholar] [CrossRef]
- Liu, M.; Li, S.; Wang, X.; Zhu, Y.; Zhang, J. Characterization, anti-oxidation and anti-inflammation of polysaccharides by Hypsizygus marmoreus against LPS-induced toxicity on lung. Int. J. Biol. Macromol. 2018, 111, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.S.; Biscaia SM, P.; Bellan, D.L.; Viana SR, F.; Carbonero, E.R. Structure elucidation of a bioactive fucomannogalactan from the edible mushroom Hypsizygus marmoreus. Carbohydr. Polym. 2019, 225, 115203. [Google Scholar] [CrossRef]
- Tsai, P.F.; Ma, C.Y. Effect of a glycoprotein from mushroom Hypsizygus marmoreus (Peck) Bigelow on growth and differentiation of human leukemic U937 cells. Food Chem. Toxicol. 2013, 62, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.H.; Tarbasa, M.; Chae, H.M.; Guan, Y.S. Molecular properties of water-unextractable proteoglycans from Hypsizygus marmoreus and their In Vitro immunomodulatory activities. Molecules 2012, 17, 207–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.G.; Kim, Y.H.; Bolormaa, Z.; Kim, M.K.; Lee, J.C. Characterization of an antihypertensive angiotensin I-converting enzyme inhibitory peptide from the edible mushroom Hypsizygus marmoreus. BioMed Res. Int. 2013, 2013, 283964. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, H.; Li, B.; Wang, X.; Liu, C.; Geng, H.; He, L. Factory-Like Bottle-Cultivation Hypsizygus marmoreus Growth Stage Carbon Dioxide Concentration Control Method. Chinese Patent CN104521559A, 22 April 2015. Available online: https://www.google.com/patents/CN104521559A?cl=en (accessed on 1 October 2021).
- Harada, A.; Yoneyama, S.; Doi, S.; Aoyama, M. Changes in contents of free amino acids and soluble carbohydrates during fruit-body development of Hypsizygus marmoreus. Food Chem. 2003, 83, 343–347. [Google Scholar] [CrossRef]
- Terashita, T.; Murao, R.; Yoshikawa, K.; Shishiyama, J. Changes in carbohydrase activities during vegetative growth and development of fruit-bodies of Hypsizygus marmoreus grown in sawdust-based culture. J. Wood Sci. 1998, 44, 234–236. [Google Scholar] [CrossRef]
- Terashita, T.; Nakaie, Y.; Inoue, T.; Yoshikawa, K.; Shishiyama, J. Role of metal proteinases in the fruit-body formation of Hypsizygus marmoreus. J. Wood Sci. 1998, 44, 379–384. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, H.; Chen, M. Cloning and functional analysis of a laccase gene during fruiting body formation in Hypsizygus marmoreus. Microbiol. Res. 2015, 179, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Nowrousian, M. Genomics and transcriptomics to study fruiting body development: An update. Fungal Biol. Rev. 2018, 32, 231–235. [Google Scholar] [CrossRef]
- Min, B.; Kim, S.; Oh, Y.L.; Kong, W.S.; Park, H.; Cho, H.; Jang, K.Y.; Kim, J.Y.; Choi, I.G. Genomic discovery of the hypsin gene and biosynthetic pathways for terpenoids in Hypsizygus marmoreus. BMC Genom. 2018, 19, 789. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, D.Y.; Park, Y.J.; Jang, M.J. Transcriptome analysis of the edible mushroom Lentinula edodes in response to blue light. PLoS ONE 2020, 15, e0230680. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ren, A.; Chen, H.; Zhao, M.; Shi, L.; Chen, M.; Wang, H.; Feng, Z.; Minou, N. Transcriptome analysis and its application in identifying genes associated with fruiting body development in basidiomycete Hypsizygus marmoreus. PLoS ONE 2015, 10, e0123025. [Google Scholar] [CrossRef] [Green Version]
- Otun, S.; Ntushelo, K. Proteomic analysis of the phytogenic fungus Sclerotinia sclerotiorum. J. Chromatogr. B 2020, 1144, 122053. [Google Scholar] [CrossRef]
- Park, Y.J.; Jang, M.J. Blue light induced edible mushroom (Lentinula edodes) proteomic analysis. J. Fungi 2020, 6, 127. [Google Scholar] [CrossRef]
- Choi, J.; Jung, W.H.; Kronstad, J.W. The cAMP/protein kinase A signaling pathway in pathogenic basidiomycete fungi: Connections with iron homeostasis. J. Microbiol. 2015, 53, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Yu, G.; Chen, Y.; Shuai, J.; Wang, M.; Jin, Y.; Lan, X.; Liang, Y.; Hui, S. Genome-wide transcriptome and proteome analysis on different developmental stages of Cordyceps militaris. PLoS ONE 2012, 7, e51853. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Guo, L.; Yu, H. Label-free comparative proteomics analysis revealed heat stress responsive mechanism in Hypsizygus marmoreus. Front. Microbiol. 2021, 11, 3359. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Manuel, B.L.; Suresh, H.; Kundu, D.J.; Avinash, I.; Johannes, G.; Gerhard, M.; Martin, E. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Van, P.; Jiang, W.; Gottardo, R.; Greg, F. ggCyto: Next generation open-source visualization software for cytometry. Bioinformatics 2018, 34, 3951–3953. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Mao, X.; Yang, J.; Xin, C.; Mao, F.; Ying, X. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef] [PubMed]
- Meijueiro, M.L.; Santoyo, F.; Ramírez, L.; Pisabarro, A.G. Transcriptome characteristics of filamentous fungi deduced using high-throughput analytical technologies. Brief. Funct. Genom. 2014, 13, 440–450. [Google Scholar] [CrossRef] [Green Version]
- Sriswasdi, S.; Takashima, M.; Manabe, R.; Ohkuma, M.; Iwasaki, W. Genome and transcriptome evolve separately in recently hybridized Trichosporon fungi. Commun. Biol. 2019, 2, 263. [Google Scholar] [CrossRef]
- Wang, G.; Chen, L.; Tang, W.; Wang, Y.; Zhang, J. Identifying a melanogenesis-related candidate gene by a high-quality genome assembly and population diversity analysis in Hypsizygus marmoreus. J. Genet. Genom. 2021, 48, 13. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, M.; Meng, J.; Feng, C.; Zhao, H.; Zhang, M. Comparative proteome reveals metabolic changes during the fruiting process in Flammulina velutipes. J. Agric. Food Chem. 2017, 65, 5091–5100. [Google Scholar] [CrossRef]
- Zhao, C.; Li, J.; Chen, N.; Bai, H.; Zhang, T. Antioxidant activity and transcriptomic analysis of Se-enriched golden oyster mushroom Pleurotus citrinopileatus (agaricomycetes). Int. J. Med. Mushrooms 2020, 22, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Hu, J.; Li, Y.; Yang, B.; Guan, Y.; Xu, C.; Chen, F.; Chi, J.; Bao, Y. Comparative proteomic analysis of Pleurotus ostreatus reveals great metabolic differences in the cap and stipe development and the potential role of ca2+ in the primordium differentiation. Int. J. Mol. Sci. 2019, 20, 6317. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wen, X.; Yang, B.; Liu, D.; Li, X.; Geng, F. De novo transcriptome and proteome analysis of Dictyophora indusiata fruiting bodies provides insights into the changes during morphological development. Int. J. Biol. Macromol. 2020, 146, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, M.; Zhang, C.; Cheng, H.; Li, T. Transcriptome and proteome analyses reveal the regulatory networks and metabolite biosynthesis pathways during the development of Tolypocladium guangdongense. Comput. Struct. Biotechnol. J. 2020, 18, 2081–2094. [Google Scholar] [CrossRef]
- Liu, J.Y.; Chang, M.; Meng, J.; Feng, C.; Liu, Y. iTRAQ-based comparative proteomics analysis of the fruiting dikaryon and the non-fruiting monokaryon of Flammulina velutipes. Curr. Microbiol. 2017, 74, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Boulton, T.G.; Yancopoulos, G.D.; Gregory, J.S.; Slaughter, C.; Moomaw, C.; Hsu, J.; Cobb, M.H. An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science 1990, 249, 64–67. [Google Scholar] [CrossRef]
- Patterson, J.C. Function and Signaling Specificity of the Hog1 Mitogen-Activated Protein Kinase in the Yeast Saccharomyces Cerevisiae; University of California: Berkeley, CA, USA, 2011. [Google Scholar]
- Duskova, M.; Borovikova, D.; Herynkova, P.; Rapoport, A.; Sychrova, H. The role of glycerol transporters in yeast cells in various physiological and stress conditions. FEMS Microbiol. Lett. 2015, 362, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Martín, H.; Rodríguez-Pachón, J.M.; Ruiz, C.; Nombela, C.; Molina, M. Regulatory mechanisms for modulation of signaling through the cell integrity Slt2-mediated pathway in Saccharomyces cerevisiae. J. Biol. Chem. 2000, 275, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.; Zhang, J.; Wang, H.; Wang, Q.; Chen, H. Comparative transcriptome analysis reveals potential fruiting body formation mechanisms in Morchella importuna. AMB Express 2019, 9, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortwendel, J.R. Orchestration of morphogenesis in filamentous fungi: Conserved roles for Ras signaling networks. Fungal Biol. Rev. 2015, 29, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Al Abdallah, Q.; Norton, T.S.; Hill, A.M.; Leclaire, L.L.; Fortwendel, J.R.; Mitchell, A.P. A fungus-specific protein domain is essential for RasA-mediated morphogenetic signaling in Aspergillus fumigatus. Msphere 2016, 1, e00234-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakafuku, M.; Obara, T.; Kaibuchi, K.; Miyajima, I.; Miyajima, A.; Itoh, H. Isolation of a second yeast Saccharomyces cerevisiae gene (GPA2) coding for guanine nucleotide-binding regulatory protein: Studies on its structure and possible functions. Proc. Natl. Acad. Sci. USA 1988, 85, 1374–1378. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Wang, Y.; Zhong, Y.; Liu, X.; Miao, M.; Duan, W. YPK1 overexpression resulted salt stress hypersensitivity in Saccharomyces cerevisiae is dependent on TOR1. Wei Sheng Wu Xue Bao = Acta Microbiol. Sin. 2009, 49, 1069–1074. [Google Scholar]
- Toda, T.; Cameron, S.; Sass, P.; Zoller, M.; Wigler, M. Cloning and characterization of BCY1, a locus encoding a regulatory subunit of the cyclic AMP-dependent protein kinase in Saccharomyces cerevisiae. Mol. Cell. Biol. 1987, 7, 1371–1377. [Google Scholar]
- Galello, F.; Moreno, S.; Rossi, S. Interacting proteins of protein kinase A regulatory subunit in Saccharomyces cerevisiae. J. Proteom. 2014, 109, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Walden, E.A.; Fong, R.Y.; Pham, T.T.; Knill, H.; Baetz, K. Phenomic screen identifies a role for the yeast lysine acetyltransferase NuA4 in the control of Bcy1 subcellular localization, glycogen biosynthesis, and mitochondrial morphology. PLoS Genet. 2020, 16, e1009220. [Google Scholar] [CrossRef]
- Almási, É.; Sahu, N.; Krizsán, K.; Bálint, B.; Kovács, G.; Kiss, B.; Cseklye, J.; Drula, E.; Henrissat, B.; Nagy, I.; et al. Comparative genomics reveals unique wood-decay strategies and fruiting body development in the Schizophyllaceae. New Phytol. 2019, 224, 902–915. [Google Scholar] [CrossRef]
- He, Z.; Luo, L.; Keyhani, N.O.; Yu, X.; Ying, S.; Zhang, Y. The c-terminal MIR-containing region in the Pmt1 o-mannosyltransferase restrains sporulation and is dispensable for virulence in Beauveria bassiana. Appl. Microbiol. Biotechnol. 2017, 101, 1143–1161. [Google Scholar] [CrossRef]
- Ban, X.; Wu, J.; Kaustubh, B.; Lahiri, P.; Li, Z. Additional salt bridges improve the thermostability of 1, 4-α-glucan branching enzyme. Food Chem. 2020, 316, 126348. [Google Scholar] [CrossRef]
- Nihira, T.; Saito, Y.; Chiku, K.; Kitaoka, M.; Ohtsubo, K.; Nakai, H. Potassium ion-dependent trehalose phosphorylase from halophilic Bacillus selenitireducens MLS10. FEBS Lett. 2013, 587, 3382–3386. [Google Scholar] [CrossRef] [Green Version]
- Macakova, E.; Kopecka, M.; Kukacka, Z.; Veisova, D.; Novak, P.; Man, P. Structural basis of the 14-3-3 protein-dependent activation of yeast neutral trehalase Nth1. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, R.; Siddalingamurthy, K.R. Co-Optimization of xyloglucanase and β-Glucosidase Using Response Surface Methodology. J. Sci. Ind. Res. 2015, 74, 450–456. [Google Scholar]
- EFSA Panel on Food Contact Materials; Enzymes and Processing Aids (CEP); Silano, V.; Barat Baviera, J.M.; Bolognesi, C.; Cocconcelli, P.; Crebelli, R.; Gott, D.; Grob, K.; Lambré, C.; et al. Safety evaluation of the food enzyme cellulase from the non-genetically modified Penicillium funiculosum strain DP-Lzc35. EFSA J. 2021, 19, e06365. [Google Scholar]
- EFSA Panel on Food Contact Materials; Enzymes and Processing Aids (CEP); Silano, V.; Barat Baviera, J.M.; Bolognesi, C.; Cocconcelli, P.; Crebelli, R.; Gott, D.; Grob, K.; Lampi, E.; et al. Safety evaluation of the food enzyme cellulase from Trichoderma reesei (strain DP-Nzc36). EFSA J. 2019, 17, e05839. [Google Scholar] [PubMed]
- Sakaguchi, M.; Makino, Y.; Matsubara, H. New approach to prepare fluorogenic branched dextrins for assaying glycogen debranching enzyme. Glycoconj. J. 2020, 37, 667–679. [Google Scholar] [CrossRef]
- Drolet, J.A. Evidence for the Involvement of the Zinc Cluster Protein Asg1p in the Transcriptional Regulation of Some Stress Response Genes in Saccharomyces cerevisiae; McGill University Libraries: Montreal, QC, Canada, 2007. [Google Scholar]
- He, X.J.; Mulford, K.E.; Fassler, J.S. Oxidative stress function of the Saccharomyces cerevisiae Skn7 receiver domain. Eukaryot. Cell 2009, 8, 768. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, A.; Ueda, J.; Yamamoto, N.; Hashikawa, N.; Sakurai, H. Role of heat shock transcription factor in Saccharomyces cerevisiae oxidative stress response. Eukaryot. Cell 2007, 6, 1373–1379. [Google Scholar] [CrossRef] [Green Version]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wang, H.; Mitchell-Silbaugh, K.; Cai, S.; Ouyang, K. Heat shock protein 60 regulates yolk sac erythropoiesis in mice. Cell Death Dis. 2019, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Morrow, G.; Tanguay, R.M. Small heat shock protein expression and functions during development. Int. J. Biochem. Cell Biol. 2012, 44, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Tichá, T.; Samakovli, D.; Kuchařová, A.; Vavrdová, T.; Šamaj, J. Multifaceted roles of HEAT SHOCK PROTEIN 90 molecular chaperones in plant development. J. Exp. Bot. 2020, 71, 3966–3985. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Lin, R.; Xu, K.; Guo, L.; Yu, H. Comparative Proteomic Analysis within the Developmental Stages of the Mushroom White Hypsizygus marmoreus. J. Fungi 2021, 7, 1064. https://doi.org/10.3390/jof7121064
Yang X, Lin R, Xu K, Guo L, Yu H. Comparative Proteomic Analysis within the Developmental Stages of the Mushroom White Hypsizygus marmoreus. Journal of Fungi. 2021; 7(12):1064. https://doi.org/10.3390/jof7121064
Chicago/Turabian StyleYang, Xiuqing, Rongmei Lin, Kang Xu, Lizhong Guo, and Hao Yu. 2021. "Comparative Proteomic Analysis within the Developmental Stages of the Mushroom White Hypsizygus marmoreus" Journal of Fungi 7, no. 12: 1064. https://doi.org/10.3390/jof7121064