
Methods for Manipulating Cryptococcus Spores


{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Producing Spores from Cryptococcus
2.1.1. Strain Selection
2.1.2. Spore Production
- (1)
- Two weeks before crossing, make large (15 cm) V8 juice agar plates: 5% V8 juice (Campbell Soup Co., Camden, NJ, USA), 2.5% Bacteriological agar (VWR International, Radnor, PA, USA, ref. no. IB49173), 0.5 g/L potassium dihydrogen phosphate, pH 7.0 (pH with 5 M potassium hydroxide). Pour plates when V8 medium is cooled to ~45 °C to minimize condensation. Allow plates to cool in short stacks (1–5 plates) at room temperature overnight. After cooling, age plates unbagged at room temperature away from direct sunlight for 14 days. After 14 days, put plates in plastic bags to prevent desiccation. Store at room temperature in the dark.
- (2)
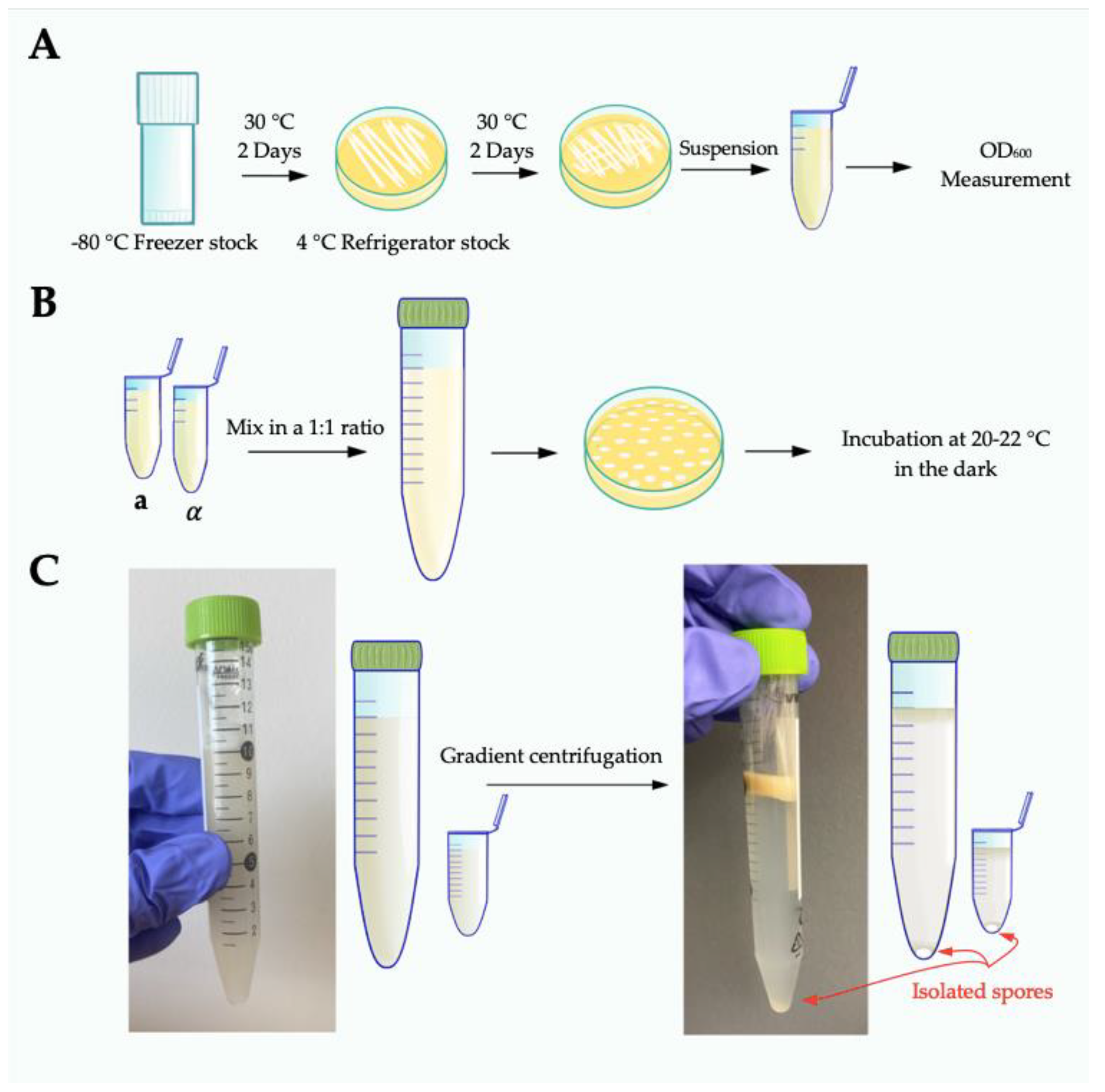
- Four days before crossing, recover strains from −80 °C freezer stocks by streaking to rich growth medium (10 cm YPD agar) and grow for 2 days at 30 °C. Re-streak sufficient cells (a mass the size of a match head) to at least half of a new 10 cm YPD plate to create a lawn. Grow for 2 days at 30 °C (Figure 1A).
- (3)
- For each strain, prepare cells by scraping up yeast from half of a 10 cm YPD plate and resuspending in 1 mL 1× phosphate buffered saline (PBS). Determine the OD600 for each resuspended strain stock. Mix strains in a 1:1 ratio in 10 mL 1× PBS to a final total OD600 equal to 2.0 (i.e., mix 1 OD600 unit JEC20 + 1 OD600 unit JEC21 and bring to a final volume of 10 mL).
- (4)
- Plate cells to 15 cm V8 plates in 10 µL spots. Maximize the distance between spots on each plate, by plating with every other tip on a multichannel pipette. Each plate can hold up to 48 spots (Figure 1B).
- (5)
- Let spots soak into agar before moving; incubate at 20–22 °C in the dark with the agar side down for 5 days.
2.2. Spore Purification Using Percoll Density Gradient Centrifugation
2.2.1. Large Scale Purification (Yield ~5 × 107 Spores/10 Plates)
- (1)
- Evaluate basidia on V8 plates for adequate spore production using a microscope. Spores are generally visible around the edge of each growth spot and can be seen in chains off the basidia at 200× magnification.
- (2)
- Prepare one 15 mL conical tube (VWR International, Radnor, PA, USA, ref no. 89039-668) per sample. One 15 mL should accommodate crosses from three to ten large (15 cm) V8 plates. Add 10 mL 75% Percoll® (GE Health Care, Chicago, IL, USA, ref no. 17-0891-01; 100% Percoll® diluted to 75% with 1× phosphate buffered saline-PBS) to each tube and place on ice. Use a cell scraper (Fisher Scientific, Hampton, NH, USA, ref no. 353086) to harvest the total mass of all spots off each V8 plate and resuspend in the Percoll®. Mix thoroughly by vortexing vigorously until the cell mass is homogenous throughout the Percoll®. It is imperative to fully homogenize the solution, as even small clumps of cells will sediment improperly during centrifugation, leading to impurities in the final spore preparations. If necessary, clumps of cells can be physically disrupted using a sterile plastic pipette tip. Keep tubes on ice throughout the harvesting process.
- (3)
- Centrifuge at 4 °C, at 2110× g, in a swinging bucket rotor for 25 min to generate a density gradient. Spores will sediment to the bottom of the tubes while all other cell types will remain at or near the top of the gradient. There should be a clear separation between spores and all other cell types (Figure 1C). If there is visible sedimentation of cells from the top of the gradient through and down to the bottom of the tube, the spores will likely not be pure.
- (4)
- Carefully remove tubes from the centrifuge and place on ice; do not disrupt the gradient.
- (5)
- Collect spores from the bottom of the density gradient:
- Gently secure the 15 mL conical tube to a ring stand, maintaining clear access to the bottom of the tube.
- Sterilize the outside of the 15 mL conical tube with 70% ethanol and dry. Remove the cap to allow the tube to vent during the next step. Venting is critical to this process; failure to do so can create a vacuum and result in sample loss.
- Use a 21-gauge needle (Fisher Scientific, Hampton, NH, USA, ref no. 1484092) to carefully and gently drill a hole in the bottom of the conical tube. It is imperative that the tube be stabilized in a manner that precludes a needle hazard. Keep both hands away from the trajectory of the needle to avoid a needle stick injury. Discard needles into a biohazard sharps container; do not recap contaminated needles!
- Immediately collect the first 200 µL that drip out in a sterile 1.5 mL microfuge tube (5–7 drops total).
- (6)
- Fill the microfuge tube to 1.5 mL with 1× PBS, vigorously pipette up and down to disperse the spores. Vortex to ensure spores are evenly suspended in solution. Centrifuge for 5 min at 2000 RPM (~370× g). Discard the top 1.3 mL of PBS, careful not to disturb the spores at the bottom. There may not be a visible pellet at this point.
- (7)
- Repeat step 6.
- (8)
- Carry out a final wash with 1.5 mL PBS and centrifuge for 1 min at maximum speed (13,200 RPM/16,100× g) to pellet spores. Discard supernatant and resuspend spore pellet in 300–700 µL PBS. Volume depends on anticipated spore yield based on visualization of spore pellet and number of V8 plates used to generate the sample. For JEC20 x JEC21 crosses after 5 days of development on V8 pH 7 agar, spore yields should be ~5 × 106 spores per V8 plate.
- (9)
- Count spores using a hemocytometer to determine final concentration and level of purity. Spores can be stored at 4 °C but begin to adhere to each other the longer they are stored.
2.2.2. Small Scale Purification (Yield ~1.5 × 106 Spores Total Per Plate)
- (1)
- Mix strains of opposite mating types together in a 1:1 ratio (OD600 = 2.0).
- (2)
- Pipet 30–40 10 µL spots on one 15 cm V8 pH 7.0 agar and incubate at 20–22 °C in the dark with the agar side down for 5–7 days.
- (3)
- Prepare one 1.5 mL microfuge tube with 1 mL 75% Percoll® and place on ice.
- (4)
- Use a cell scraper to scrape the total mass of cells and resuspend in the 75% Percoll®. Resuspend thoroughly by vigorous vortexing until the cell mass appears homogenous throughout. If any clumps are still visible, pipette up and down to remove them. Keep tubes on ice throughout harvest of spots.
- (5)
- Centrifuge at 4 °C at 2000× g for 25 min to generate a density gradient. Spores will sediment to the bottom of the tubes. All other cell types will remain at the top of the gradient. There will be a clear separation between spores and other cells. If there is visible sedimentation of cells from the top of the gradient down to the bottom of the tube, the spores will not be pure.
- (6)
- Carefully remove tubes from the centrifuge and place on ice.
- (7)
- Collect spores from the bottom of the gradient:
- Open the lid of the 1.5 mL gradient tube. Sterilize the bottom with 70% Ethanol.
- Use a 23-gauge needle to carefully and gently drill a hole in the bottom of the tube.
- Immediately collect the first 50–100 μL that drips out in a sterile 1.5 mL microfuge tube (2–3 drops total).
- (8)
- Fill the microfuge tube to 1.5 mL with 1× PBS, pipetting up and down to disperse the spores. Vortex to ensure spores are evenly suspended in solution. Centrifuge for 5 min at 2000 RPM (~370× g). Discard the top 1.3 mL of PBS, careful not to disturb the spores at the bottom. There may not be a pellet after the first two spins.
- (9)
- Repeat step 8.
- (10)
- Carry out a final wash with 1.5 mL PBS and centrifuge for 1 min at maximum speed (13,200 RPM/16,100× g). Discard supernatant and resuspend spore pellet in ~50 µL PBS per 5 spots purified (can be adjusted according to size of spore pellet).
- (11)
- Count spores using a hemocytometer to determine final concentration and purity. Spore yield should be ~1 × 106 per 30 spots.
3. Quantitative Germination Assays (QGAs)
Microscopy-Based Quantitative Germination Assay
- (1)
- The microscope and stage-top incubator need to be preheated to minimize thermal drift during long-term experiments. At least 1 h before starting the experiment, fill the stage-top incubator with dH2O, place the heating ring on the desired objective (20×) and turn the heating element on to 30 °C.
- (2)
- Prepare a 384-well plate (Thermo Fisher Scientific, Waltham, MA, USA, ref no. 142762) with spores, allow them to settle, then add 2× germination medium just prior to starting the assay as follows:
- Dilute highly pure (>97%) spores to 5000 spores/μL in 1× PBS. Add 20 μL of spores to each well (100,000 spores/well). Be sure not to touch the pipette tip to the bottom of the well as this can lead to scratches that interfere with imaging. Allow spores to settle and adhere to the bottom of the well for ~15 min at 4 °C prior to addition of media. Prepare each sample in triplicate wells.
- Add 20 μL 2× germination media to each well, pipetting up and down gently 5 times to mix. Synthetic Medium with dextrose (2× Synthetic Minimal Medium, 4% Dextrose) is a defined medium that promotes synchronous and efficient germination.
- Cover plate with plastic wrap to prevent evaporation during the experiment. Place the plastic plate cover on top of the plastic.
- Place the 384-well plate in the stage-top incubator, ensuring it is pushed fully down and sitting flat on the microscope stage.
- (3)
- Using the 20× objective (Nikon CFI Plan Apochromat Lambda 20XC), maneuver to the location of the first well. Make sure to enable the perfect focus (PFS) on the microscope. Spores are in focus when they are dark ovals without any halo-ed edges. Select X, Y, and Z coordinates for each position. Make sure that there is no overlap between images. Select three images per well for a total of nine images per sample.
- (4)
- Once all image locations have been selected, inspect each image individually, and reset the Z coordinate so that each image is in focus, as thermal drift may occur as the experiment is being set up.
- (5)
- Set up Z-stack imaging ranging from +7.5 μm to −7.5 μm from your selected Z-coordinates, with imaging every 1.5 μm for a total of 21 images per position. Z-stacks ensure that in-focus images will be obtained across the time course of germination, regardless of any thermal drift. This step is essential because the PFS is not designed to focus on many small cells, as is done in this assay.
- (6)
- Set the microscope to take images every 2 h for 16 h for a total of 9 timepoints. This is the standard imaging time frame for wild type spore germination in SD medium, although longer time courses and alternative intervals can be performed as needed for individual experiments.
- (7)
- Ensure all desired parameters are properly set for the automated experiment (time loop, each XYZ positions, Z-stack, etc.) before starting the image acquisition. Initiate image acquisition.
- (8)
- Once the image collection is completed, export the images to TIFF files.
- (9)
- Select the image from the Z-stack that is most in-focus for each position and time point. Spores should be dark (black/gray) and free of haloed edges.
- (10)
- Place images in corresponding folders with the following progression/experiment/sample/time (0H, 2H, etc.)/Input. Create an “output” and “data” folder under each time point before running ImageJ.
- (11)
- Run each sample folder through the ImageJ program (File S1). This program quantifies the area and aspect ratio of each spot in the image and compiles them into a spreadsheet.
- (12)
- Run each sample folder through the MATLAB program (File S2). This program determines how many spots fit “spore,” “yeast,” or “other” parameters based on size and aspect ratio. It outputs a 2D histogram with size on the X-axis and aspect ratio on the Y-axis for each time point. Population level changes can be tracked over time.
- (13)
- This program also outputs a data.csv file, which can be used for further data manipulation (creating bar plots and rate curves).
4. Molecular Methods
4.1. RNA Isolation from Spores
- (1)
- Pellet purified spores by centrifuging for 5 min at 10,000 RPM (~9200× g) at 4 °C.
- (2)
- Discard supernatant and resuspend the pellet in 1 mL ice-cold water. Transfer to a clean microfuge. Centrifuge at 13,200 RPM/16,100× g for 2 min at 4 °C and remove supernatant.
- (3)
- Resuspend pellet in 0.4 mL TES buffer (50 mM Tris-HCl pH 7.5, 10 mM EDTA, 1% SDS). Add 0.4 mL 65 °C acid phenol:chloroform (Fisher Scientific, Hampton, NH, USA, ref no. AM9720) and vortex vigorously 10 for seconds. Incubate for 60 min at 65 °C with brief mixing every 10 min (do not vortex).
- (4)
- Place on ice for 5 min. Centrifuge 5 min at 13,200 RPM/16,100× g at 4 °C.
- (5)
- Transfer the top aqueous phase to a clean microfuge tube, add 0.4 mL chloroform, and vortex vigorously. Place on ice for 5 min and centrifuge at maximum speed at 4 °C.
- (6)
- Transfer aqueous phase to a clean microfuge tube and add 0.4 mL chloroform. Vortex vigorously and centrifuge 5 min at maximum speed at 4 °C.
- (7)
- Transfer aqueous phase to a new tube and precipitate the RNA by adding 0.1× volume of 3 M sodium acetate pH 5.3 (Fisher Scientific, Hampton, NH, USA ref no. R1181) (i.e., 40 μL for 400 μL aqueous phase) and 2.5× volume of ice-cold 100% ethanol. For very small amounts of RNA, add 20 μg of RNase-free glycogen (Thermo Fisher Scientific, Waltham, MA, USA, ref no. R0551) to facilitate precipitation.
- (8)
- Centrifuge 30 min at 13,200 RPM/16,100× g at 4 °C to pellet precipitating RNA. Remove supernatant. Wash RNA pellet by vortexing briefly in 0.5 mL ice-cold 70% ethanol. Centrifuge 5 min at top speed at 4 °C.
- (9)
- Remove ethanol from the pellet and evaporate remaining ethanol from the pellet. Do not over dry the pellet. EITHER:Place tubes upside down and allow ethanol to drain for 20 min, ORPlace open tubes in 65 °C heat block or water bath for 10 min.
- (10)
- Resuspend pellet in 30–50 μL RNase-free water. Vortex vigorously to ensure that the colorless pellet is fully resuspended. Determine the RNA concentration and purity spectrophotometrically by measuring the A260 and A280. Store at −80 °C or at −20 °C, if it is to be used within 1 year.
4.2. Protein Isolation from Spores
- (1)
- Pellet purified spores by centrifuging for 5 min at 10,000 RPM (~9200× g) at 4 °C.
- (2)
- Resuspend spore pellet in 500 μL TES buffer (50 mM Tris-HCl pH 7.5, 10 mM EDTA, 1% SDS), and sonicate with a microtip 5 times for 12 s each at power 2 and a 100% duty cycle (VWR International, Radnor, PA, USA, model no. 101-135-022). Cool on ice for 1 min between cycles to avoid overheating the sample.
- (3)
- Add 200 μL phenol-chloroform (Fisher Scientific, Hampton, NH, USA, ref no. 0883-400ML) and mix well by vortexing. Centrifuge at maximum speed (13,200 RPM/16,100× g) for 2 min. Discard top aqueous layer, add 1 mL 100% ethanol, and mix well.
- (4)
- Centrifuge at maximum speed for 2 min. Discard supernatant and wash pellet with 500 μL 100% ethanol and 500 μL acetone.
- (5)
- Discard supernatant, and dry pellet in SpeedVac for 10–15 min. The resulting protein pellet contains ~1 µg of protein/1 × 109 spores and can be resuspended in 100 µL SDS PAGE loading buffer (0.025 M Tris–HCl pH 8.3, 0.192 M glycine, 0.1% SDS) and boiled for 5 min for subsequent electrophoretic separation or undergo processing for other, non-gel-based analyses.
5. Host–Pathogen Interactions
5.1. In Vitro Spore Phagocytosis Assays
5.1.1. Colony Forming Unit Association Assay
- (1)
- Seed phagocytes in a sterile µ-slide 8-well coverslip/plate (Ibidi, Fitchburg, WI, USA, ref no. 80826) at a density of 2.5 × 105 cells/mL in a volume of 200 µL per well. Incubate overnight to allow for settling and adhesion.
- (2)
- Add spores at an MOI of 10:1 and allow phagocytosis to occur for 4 h.
- (3)
- Wash three times with 1× phosphate buffered saline (PBS) to remove spores not adhered to phagocytes.
- (4)
- Add 300 µL 0.01% Triton X-100 to each well to lyse the phagocytes. (This concentration is known to lyse mammalian phagocytes but not affect the viability of spores.)
- (5)
- Serially dilute the lysate by 10-fold 3 times and plate each sample on a yeast peptone dextrose (YPD) agar plate. Incubate each dilution at 30 °C for 3 days and count visible colonies. Percent phagocyte association is calculated by dividing the number of colonies by the number of spores added to the phagocytes (100 × (# CFUs from lysate/CFUs introduced to phagocytes)).
5.1.2. Fluorescence-Based Internalization Assay
- (1)
- Seed phagocytic cells in a 96-well plate with ~3 × 104 cells in 100 µL RPMI + 10% fetal bovine serum (FBS). Incubate cells overnight at 37 °C in 5% CO2 to allow for cell adhesion.
- (2)
- Pellet purified spores by centrifuging for 5 min at 10,000 RPM (~9200× g) at 4 °C. Remove supernatant and fix spores by incubating in 4% formaldehyde overnight at 4 °C.
- (3)
- Wash spores with 1× phosphate buffered saline (PBS), remove supernatant. Resuspend in 1× PBS and stain with 0.1 mg/mL calcofluor white (Sigma-Aldrich, St. Louis, MO, USA, ref no. 18909-100ML-F). Incubate 5–30 min at ambient temperature in the dark. Wash with 1× PBS 3 times.
- (4)
- Remove medium from phagocytes. Add spores at an MOI of 100:1 (3 × 106 spores/well) in 100 µL fresh RPMI + FBS. Incubate 4 h at 37 °C to allow for measurable phagocytosis.
- (5)
- Measure the fluorescence of each well with a plate reader at 347 nm to determine the amount of spore-associated fluorescence in each well.
- (6)
- Add 100 µL 0.4% trypan blue to quench fluorescence of spores that were not phagocytosed. Measure fluorescence at 347 nm of each well with a plate reader again.
- (7)
- Generate a 10-fold serial dilution control curve to determine the amount of fluorescence per spore for calcofluor white (total). Calculate percent phagocytosis by dividing the number of spores phagocytosed by the number of spores added to the phagocytes and multiplying by 100 (100 × (no. of spores phagocytosed/no. of spores introduced)).
5.2. Animal Models of Infection with Spores
Murine Models
- (1)
- Prepare spores in 1× phosphate buffered saline (PBS). Count spores using a hemacytometer and prepare appropriate samples for inoculation. An effective inoculum of spores from most strain pairs is 1 × 105 to 5 × 105 spores per mouse. Dilute spores in enough volume of PBS for 50 µL inoculum per mouse plus an extra 10 µL for post-infection plating to confirm CFUs introduced into the mice.
- (2)
- Place mice in a SurgiVet chamber to anesthetize with isoflurane. Set the oxygen flow rate to 2 L/minute and the isoflurane setting to 4. Maintain anesthesia throughout inoculation procedure.
- (3)
- Suspend up to 5 anesthetized mice by their incisors on a silk thread so that the necks are fully extended.
- (4)
- Slowly pipette half of the 50 µL of spore suspension directly into each nostril one drop at a time, ensuring that the mice fully inhale each drop. Allow the mice to remain suspended under anesthesia for 10 min after infection.
- (5)
- After 10 min, remove mice from anesthesia chamber. Monitor mice for 10 to 15 min after they regain consciousness to ensure full recovery.
- (6)
- Dilute remaining 10 µL inoculum to 1 spore/µL in 1× PBS and plate 100–200 µL on a yeast peptone dextrose (YPD) agar plate. Incubate for 3 days at 30 °C. Calculate the number of CFUs of the inoculum to determine infectious dose that was administered.
- (7)
- Continue monitoring mice twice per day for 30–100 days, depending on parental strain pair used to generate spores. Check mice for signs of disease such as labored breathing, hunching, squinting, brain swelling, or paralysis. Euthanize any animals showing signs of distress or disease immediately upon discovery.
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Driks, A. Overview: Development in Bacteria: Spore Formation in Bacillus Subtilis. Cell. Mol. Life Sci. (CMLS) 2002, 59, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Kessin, R.H. Dictyostelium: Evolution, Cell Biology, and the Development of Multicellularity; Developmental and Cell Biology Series; Cambridge University Press: Cambridge, UK, 2001; ISBN 978-0-521-58364-0. [Google Scholar]
- Wyatt, T.T.; Wösten, H.A.B.; Dijksterhuis, J. Fungal Spores for Dispersion in Space and Time. In Advances in Applied Microbiology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 85, pp. 43–91. ISBN 978-0-12-407672-3. [Google Scholar]
- Neiman, A.M. Sporulation in the Budding Yeast Saccharomyces Cerevisiae. Genetics 2011, 189, 737–765. [Google Scholar] [CrossRef] [Green Version]
- Adams, T.H.; Wieser, J.K.; Yu, J.H. Asexual Sporulation in Aspergillus Nidulans. Microbiol. Mol. Biol. Rev. 1998, 62, 35–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.-W.; Lee, K.-T.; So, Y.-S.; Bahn, Y.-S. Genetic Manipulation of Cryptococcus neoformans. Curr. Protoc. Microbiol. 2018, 50, e59. [Google Scholar] [CrossRef] [PubMed]
- Kwon-Chung, K.J. A New Genus, Filobasidiella, the Perfect State of Cryptococcus neoformans. Mycologia 1975, 67, 1197. [Google Scholar] [CrossRef]
- Kwon-Chung, K.J. Morphogenesis of Filobasidiella neoformans, the Sexual State of Cryptococcus neoformans. Mycologia 1976, 68, 821. [Google Scholar] [CrossRef]
- Lin, X.; Hull, C.M.; Heitman, J. Sexual Reproduction between Partners of the Same Mating Type in Cryptococcus neoformans. Nature 2005, 434, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Priest, S.J.; Heitman, J. Cryptococcus neoformans Mating and Genetic Crosses. Curr. Protoc. Microbiol. 2019, 53, e75. [Google Scholar] [CrossRef]
- Botts, M.R.; Giles, S.S.; Gates, M.A.; Kozel, T.R.; Hull, C.M. Isolation and Characterization of Cryptococcus neoformans Spores Reveal a Critical Role for Capsule Biosynthesis Genes in Spore Biogenesis. Eukaryot. Cell 2009, 8, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Velagapudi, R.; Hsueh, Y.-P.; Geunes-Boyer, S.; Wright, J.R.; Heitman, J. Spores as Infectious Propagules of Cryptococcus neoformans. Infect. Immun. 2009, 77, 4345–4355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, N.M.; Botts, M.R.; McDermott, A.J.; Ortiz, S.C.; Wüthrich, M.; Klein, B.; Hull, C.M. Infectious Particle Identity Determines Dissemination and Disease Outcome for the Inhaled Human Fungal Pathogen Cryptococcus. PLoS Pathog 2019, 15, e1007777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, S.C.; Huang, M.; Hull, C.M. Discovery of Fungus-Specific Targets and Inhibitors Using Chemical Phenotyping of Pathogenic Spore Germination. mBio 2021, 12, e0167221. [Google Scholar] [CrossRef] [PubMed]
- Kwon-Chung, K.J.; Edman, J.C.; Wickes, B.L. Genetic Association of Mating Types and Virulence in Cryptococcus neoformans. Infect. Immun. 1992, 60, 602–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadwick, B.J.; Lin, X. On the History and Applications of Congenic Strains in Cryptococcus Research. Pathogens 2020, 9, 750. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J.; Allen, B.; Alspaugh, J.A.; Kwon-Chung, K.J. On the Origins of Congenic MATα and MATa Strains of the Pathogenic Yeast Cryptococcus neoformans. Fungal Genet. Biol. 1999, 28, 1–5. [Google Scholar] [CrossRef]
- Toffaletti, D.L.; Nielsen, K.; Dietrich, F.; Heitman, J.; Perfect, J.R. Cryptococcus neoformans Mitochondrial Genomes from Serotype A and D Strains Do Not Influence Virulence. Curr. Genet. 2004, 46, 193–204. [Google Scholar] [CrossRef]
- Giles, S.S.; Dagenais, T.R.T.; Botts, M.R.; Keller, N.P.; Hull, C.M. Elucidating the Pathogenesis of Spores from the Human Fungal Pathogen Cryptococcus neoformans. Infect. Immun. 2009, 77, 3491–3500. [Google Scholar] [CrossRef] [Green Version]
- Litvintseva, A.P.; Marra, R.E.; Nielsen, K.; Heitman, J.; Vilgalys, R.; Mitchell, T.G. Evidence of Sexual Recombination among Cryptococcus neoformans Serotype A Isolates in Sub-Saharan Africa. Eukaryot. Cell 2003, 2, 1162–1168. [Google Scholar] [CrossRef] [Green Version]
- Springer, D.J.; Phadke, S.; Billmyre, B.; Heitman, J. Cryptococcus gattii, No Longer an Accidental Pathogen? Curr. Fungal Infect. Rep. 2012, 6, 245–256. [Google Scholar] [CrossRef]
- Juhos, E.T. Density Gradient Centrifugation of Bacteria and Nonspecific Bacteriophage in Silica Sol. J. Bacteriol. 1966, 91, 1376–1377. [Google Scholar] [CrossRef] [Green Version]
- Blaske-Lietze, V.-U.; Shapiro, A.M.; Denton, J.S.; Botts, M.; Becnel, J.J.; Boucias, D.G. Development of the Insect Pathogenic Alga Helicosporidium. J. Eukaryot. Microbiol. 2006, 53, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Barkal, L.J.; Walsh, N.M.; Botts, M.R.; Beebe, D.J.; Hull, C.M. Leveraging a High Resolution Microfluidic Assay Reveals Insights into Pathogenic Fungal Spore Germination. Integr. Biol. 2016, 8, 603–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Hebert, A.S.; Coon, J.J.; Hull, C.M. Protein Composition of Infectious Spores Reveals Novel Sexual Development and Germination Factors in Cryptococcus. PLoS Genet. 2015, 11, e1005490. [Google Scholar] [CrossRef] [Green Version]
- Chun, C.D.; Brown, J.C.S.; Madhani, H.D. A Major Role for Capsule-Independent Phagocytosis-Inhibitory Mechanisms in Mammalian Infection by Cryptococcus neoformans. Cell Host Microbe 2011, 9, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leopold Wager, C.M.; Hole, C.R.; Wozniak, K.L.; Wormley, F.L. Cryptococcus and Phagocytes: Complex Interactions That Influence Disease Outcome. Front. Microbiol. 2016, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Huang, M.; Botts, M.R.; Hull, C.M.; Huttenlocher, A. A Zebrafish Model of Cryptococcal Infection Reveals Roles for Macrophages, Endothelial Cells, and Neutrophils in the Establishment and Control of Sustained Fungemia. Infect. Immun. 2016, 84, 3047–3062. [Google Scholar] [CrossRef] [Green Version]
- Solotorovsky, M.; Bugie, E.J. The Effect of Streptothricin on Systemic Infection with Cryptococcus neoformans in Mice. J. Immunol. 1948, 60, 497–502. [Google Scholar] [PubMed]
- Sukroongreung, S.; Kitiniyom, K.; Nilakul, C.; Tantimavanich, S. Pathogenicity of Basidiospores of Filobasidiella neoformans var. neoformans. Med. Mycol. 1998, 36, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, B.L.; Hempel, H.O.; Goodman, N.L. Pathogenicity of the Basidiospores of Filobasidiella neoformans. Mycopathologia 1984, 85, 149–153. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frerichs, A.B.; Huang, M.; Ortiz, S.C.; Hull, C.M. Methods for Manipulating Cryptococcus Spores. J. Fungi 2022, 8, 4. https://doi.org/10.3390/jof8010004
Frerichs AB, Huang M, Ortiz SC, Hull CM. Methods for Manipulating Cryptococcus Spores. Journal of Fungi. 2022; 8(1):4. https://doi.org/10.3390/jof8010004
Chicago/Turabian StyleFrerichs, Anna B., Mingwei Huang, Sébastien C. Ortiz, and Christina M. Hull. 2022. "Methods for Manipulating Cryptococcus Spores" Journal of Fungi 8, no. 1: 4. https://doi.org/10.3390/jof8010004
APA StyleFrerichs, A. B., Huang, M., Ortiz, S. C., & Hull, C. M. (2022). Methods for Manipulating Cryptococcus Spores. Journal of Fungi, 8(1), 4. https://doi.org/10.3390/jof8010004