
Whole-Genome Sequencing and Transcriptome Analysis of Ganoderma lucidum Strain Yw-1-5 Provides New Insights into the Enhanced Effect of Tween80 on Exopolysaccharide Production
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain and Mycelium Nucleic Acid Preparation
2.2. De Novo Sequencing and Assembly
2.3. Genomic Component Analysis and Genome Annotation
2.4. Tween80 Treatment and Transcriptome Analysis
2.5. Real-Time Quantitative PCR
2.6. Data Availability
3. Results
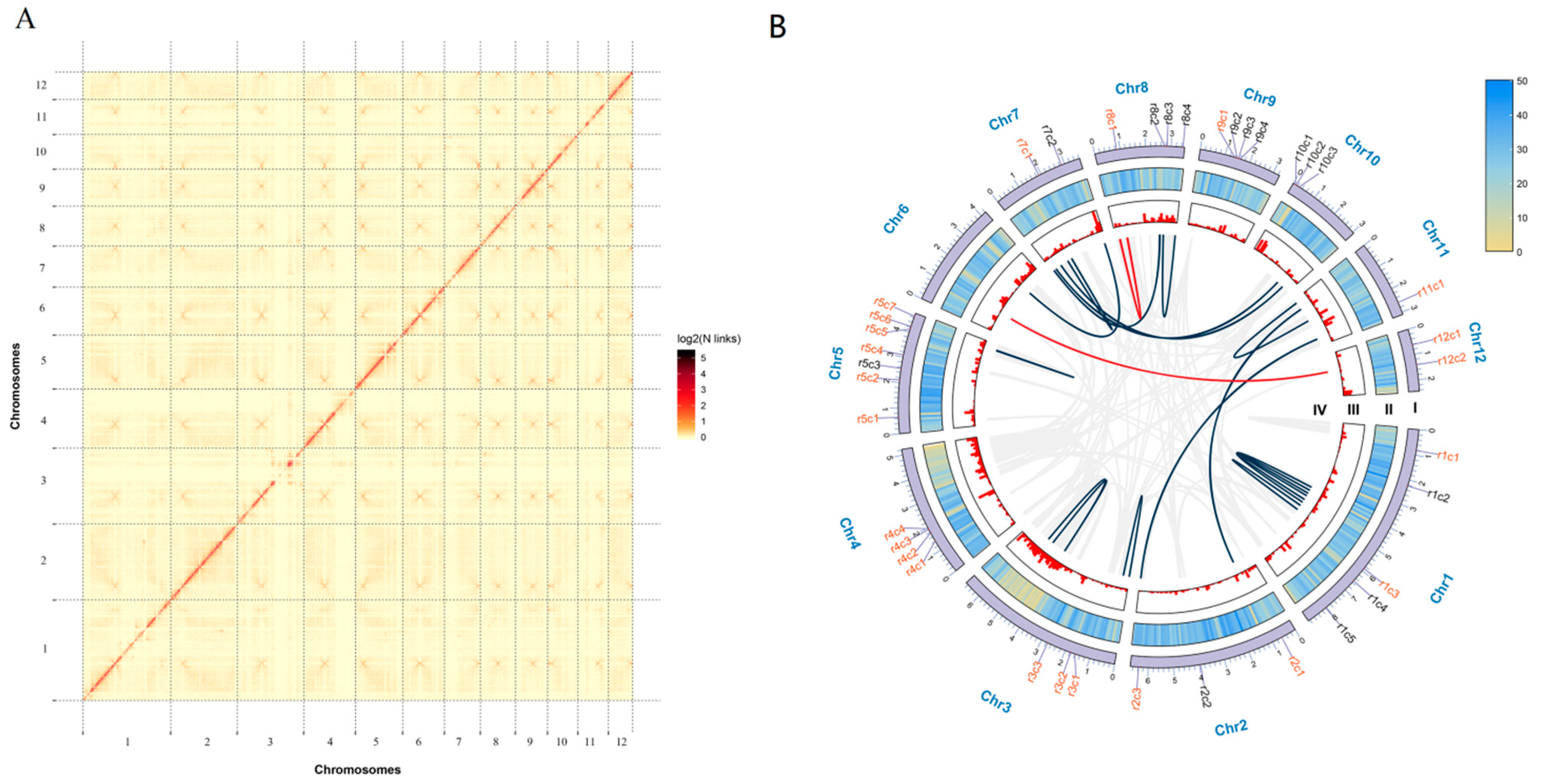
3.1. Sequencing and De Novo Assembly of Ganoderma lucidum Genome
3.2. Gene Prediction of Ganoderma lucidum Yw-1-5 Genome
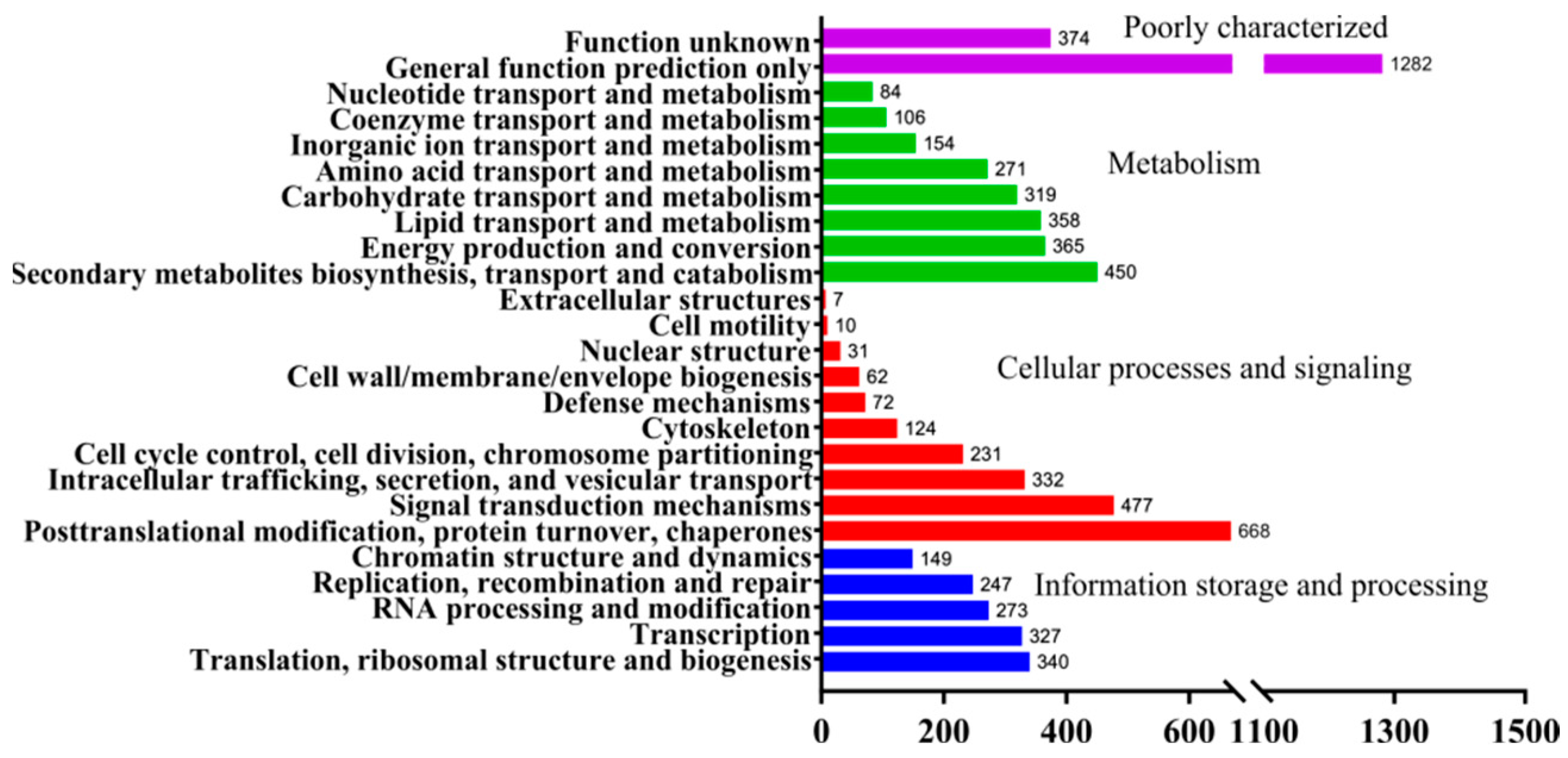
3.3. Functional Annotation of Ganoderma lucidum Yw-1-5 Genome
3.4. Annotation in CAZy Databases
3.5. Genes Involved in Synthesis of Polysaccharides from G. lucidum Genome
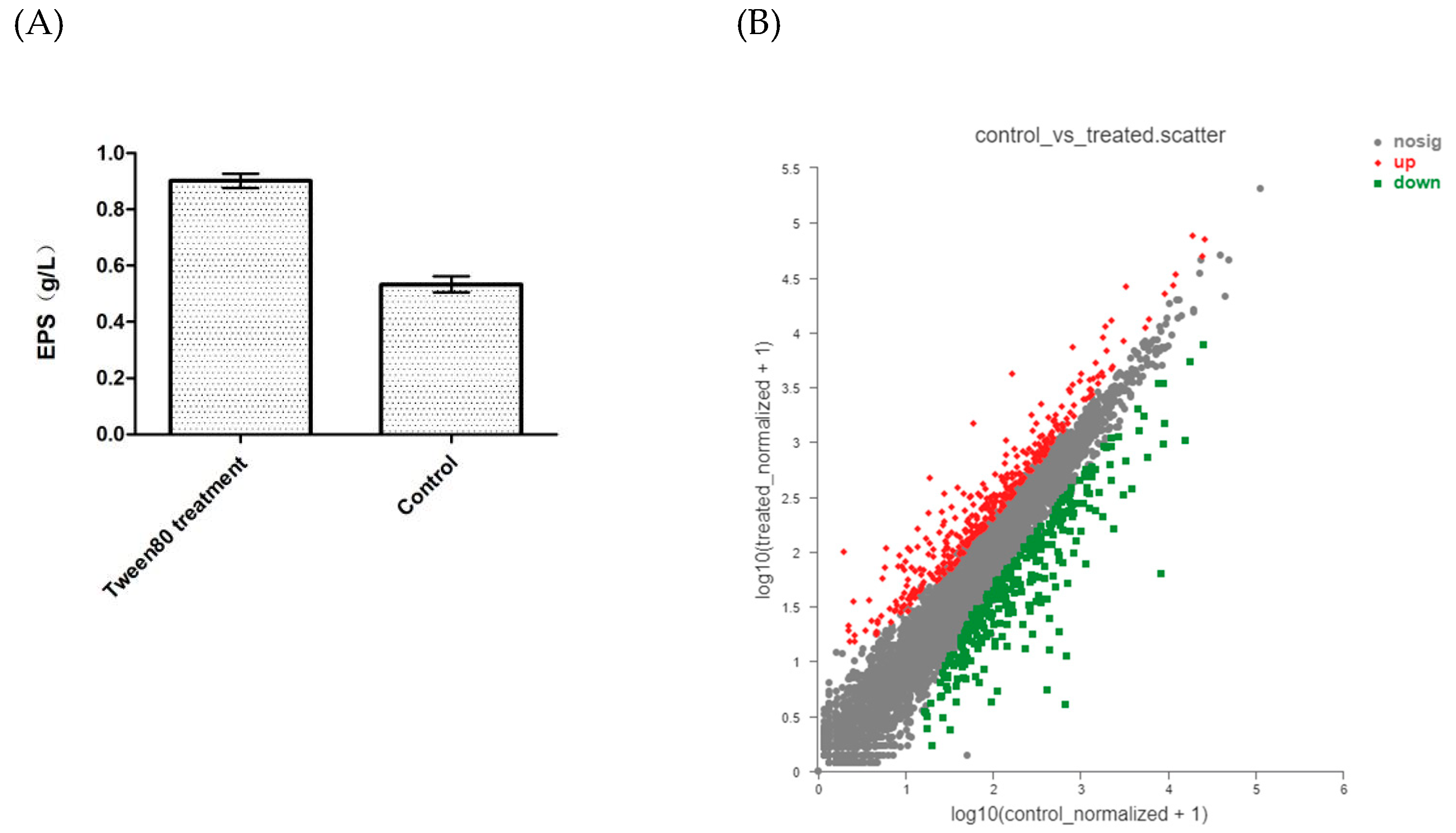
3.6. Transcriptomic Analysis of G. lucidum Mycelium Treated with Tween80
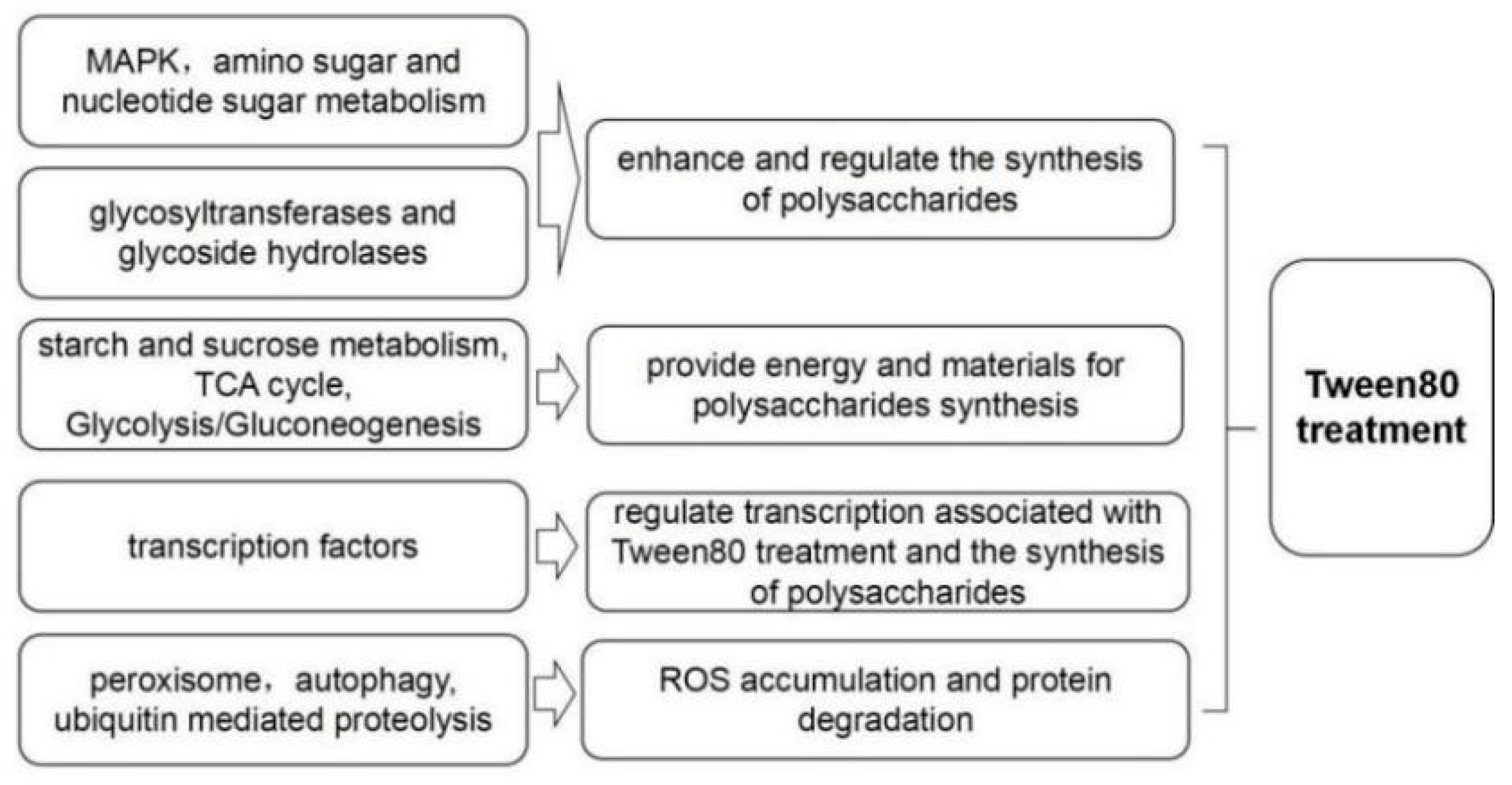
3.7. Differentially Expressed Genes Involved in the Enhanced Effect of Tween80 on Exopolysaccharide Production
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, K.; Liu, Y.; Zhao, X.; Tang, Q.; Dernedde, J.; Zhang, J.; Fan, H. Anti-inflammatory properties of GLPss58, a sulfated polysaccharide from Ganoderma lucidum. Int. J. Biol. Macromol. 2018, 107, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Wu, Q.; Xie, Y.; Tan, J.; Ding, Y.; Bai, L. Hypoglycemic mechanisms of Ganoderma lucidum polysaccharides F31 in db/db mice via RNA-seq and iTRAQ. Food Funct. 2018, 9, 6495–6507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, H.; Song, L.; Xue, J.; Wang, X.; Song, S.; Wang, S. Polysaccharide from Ganoderma lucidum ameliorates cognitive impairment by regulating the inflammation of the brain-liver axis in rats. Food Funct. 2021, 12, 6900–6914. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; He, R.; Sun, P.; Zhang, F.; Linhardt, R.J.; Zhang, A. Molecular mechanisms of bioactive polysaccharides from Ganoderma lucidum (Lingzhi), a review. Int. J. Biol. Macromol. 2020, 150, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Jan, J.; Cheng, T.R.; Juang, Y.; Ma, H.; Wu, Y.; Yang, W.; Cheng, C.; Chen, X.; Chou, T.; Shie, J.; et al. Identification of existing pharmaceuticals and herbal medicines as inhibitors of SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2021579118. [Google Scholar] [CrossRef]
- Papinutti, L. Effects of nutrients, pH and water potential on exopolysaccharides production by a fungal strain belonging to Ganoderma lucidum complex. Bioresour Technol. 2010, 101, 1941–1946. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, G.; Huang, F.; Wu, X.; Yang, H. Improved production, purification and bioactivity of a polysaccharide from submerged cultured Ganoderma lucidum. Arch. Pharmacal Res. 2014, 37, 1530–1537. [Google Scholar] [CrossRef]
- Zhang, W.; Tang, Y.-J. A Novel Three-Stage Light Irradiation Strategy in the Submerged Fermentation of Medicinal Mushroom Ganoderma lucidum for the Efficient Production of Ganoderic Acid and Ganoderma Polysaccharides. Biotechnol. Prog. 2008, 24, 1249–1261. [Google Scholar] [CrossRef]
- Liang, Y.; Zhu, L.; Gao, M.; Zheng, Z.; Wu, J.; Zhan, X. Influence of Tween-80 on the production and structure of water-insoluble curdlan from Agrobacterium sp. Int. J. Biol. Macromol. 2018, 106, 611–619. [Google Scholar] [CrossRef]
- Li, Q.; Lei, Y.; Hu, G.; Lei, Y.; Dan, D. Effects of Tween 80 on the liquid fermentation of Lentinus edodes. Food Sci. Biotechnol. 2018, 27, 1103–1109. [Google Scholar] [CrossRef]
- Meng, Q.; Chuai, S.; Chen, L.; Wang, L.; Cai, G.; Mao, J.; Gu, Z.; Shi, G.; Ding, Z. Effect of surfactants on the production of polysaccharides from Schizophyllum commune through submerged fermentation. Int. J. Biol. Macromol. 2021, 192, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Zhang, Y.; He, J.; Xie, Y. Enhanced exopolysaccharide production in submerged fermentation of Ganoderma lucidum by Tween 80 supplementation. Bioprocess Biosyst. Eng. 2021, 44, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Schmid, J.; Sieber, V. Enzymatic transformations involved in the biosynthesis of microbial exo-polysaccharides based on the assembly of repeat units. Chembiochem 2015, 16, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-W.; Ji, S.-L.; Li, H.-J.; Zhou, J.-S.; Duan, Y.-Q.; Dang, L.-Z.; Mo, M.-H. Increased polysaccharide production and biosynthetic gene expressions in a submerged culture of Ganoderma lucidum by the overexpression of the homologous α-phosphoglucomutase gene. Bioprocess Biosyst. Eng. 2015, 38, 399–405. [Google Scholar] [CrossRef]
- Li, M.; Chen, T.; Gao, T.; Miao, Z.; Jiang, A.; Shi, L.; Ren, A.; Zhao, M. UDP-glucose pyrophosphorylase influences polysaccharide synthesis, cell wall components, and hyphal branching in Ganoderma lucidum via regulation of the balance between glucose-1-phosphate and UDP-glucose. Fungal Genet. Biol. 2015, 82, 251–263. [Google Scholar] [CrossRef]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [Green Version]
- Thanh Nguyen, H.; Zhang, R.; Inokawa, N.; Oura, T.; Chen, X.; Iwatani, S.; Niimi, K.; Niimi, M.; Holmes, A.R.; Cannon, R.D.; et al. Candida albicans Bgl2p, Ecm33p, and Als1p proteins are involved in adhesion to saliva-coated hydroxyapatite. J. Oral Microbiol. 2021, 13, 1879497. [Google Scholar] [CrossRef]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.-Z.; Wang, Z.-F.; Liu, Y.-D.; Zhang, G.-Z.; Li, G. The whole-genome sequencing and analysis of a Ganoderma lucidum strain provide insights into the genetic basis of its high triterpene content. Genomics 2021, 113, 840–849. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, L.; Shang, X.; Peng, B.; Li, Y.; Xiao, S.; Tan, Q.; Fu, Y. Chromosomal genome and population genetic analyses to reveal genetic architecture, breeding history and genes related to cadmium accumulation in Lentinula edodes. BMC Genom. 2022, 23, 120. [Google Scholar] [CrossRef]
- Yang, L.; Tang, J.; Chen, J.-J.; Peng, A.-Y.; Wang, Q.-M.; Rao, L.-Q.; Yang, H.; Zhang, X.-W.; Yang, H.-Z.; Zhang, C.; et al. Transcriptome analysis of three cultivars of Poria cocos reveals genes related to the biosynthesis of polysaccharides. J. Asian Nat. Prod. Res. 2019, 21, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Tang, Z.; Zhang, J.; Li, X.; Yang, Z.; Yang, C.; Zhang, Z.; Huang, Z. Comparative transcriptome analysis reveals the genetic basis underlying the biosynthesis of polysaccharides in Hericium erinaceus. Bot. Stud. 2019, 60, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Chen, J.; Liu, J.; Yu, H.; Zhang, L.; Song, C.; Li, Y.; Jiang, N.; Tan, Q.; Shang, X.; et al. De novo Sequencing and Comparative Transcriptome Analyses Provide First Insights Into Polysaccharide Biosynthesis During Fruiting Body Development of Lentinula edodes. Front. Microbiol. 2021, 12, 627099. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Belton, J.-M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi—C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Wessler, S.R. MITE-Hunter: A program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010, 38, e199. [Google Scholar] [CrossRef] [Green Version]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21 (Suppl. S1), i351–i358. [Google Scholar] [CrossRef]
- Edgar, R.C.; Myers, E.W. PILER: Identification and classification of genomic repeats. Bioinformatics 2005, 21 (Suppl. S1), i152–i158. [Google Scholar] [CrossRef] [PubMed]
- Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19 (Suppl. S2), i215–i225. [Google Scholar] [CrossRef] [Green Version]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef] [Green Version]
- Blanco, E.; Parra, G.; Guigó, R. Using geneid to identify genes. Curr. Protoc. Bioinform. 2007, 18, 3–4. [Google Scholar]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef] [PubMed]
- Sirén, J.; Välimäki, N.; Mäkinen, V. Indexing Graphs for Path Queries with Applications in Genome Research. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.J.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramírez, F.; Vesztrocy, A.W.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.-Y.J.; Fan, W.-L.; Wang, W.-F.; Chen, T.; Tang, Y.-C.; Chu, F.-H.; Chang, T.-T.; Wang, S.-Y.; Li, M.-Y.; Chen, Y.-H.; et al. Genomic and transcriptomic analyses of the medicinal fungus Antrodia cinnamomea for its metabolite biosynthesis and sexual development. Proc. Natl. Acad. Sci. USA 2014, 111, E4743–E4752. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Huang, L.; Hu, H.; Cai, M.; Liang, X.; Li, X.; Zhang, Z.; Xie, Y.; Xiao, C.; Chen, S.; et al. Whole-genome assembly of Ganoderma leucocontextum (Ganodermataceae, Fungi) discovered from the Tibetan Plateau of China. G3 Genes Genomes Genet. 2021, 11, jkab337. [Google Scholar] [CrossRef]
- González-Rubio, G.; Sastre-Vergara, L.; Molina, M.; Martín, H.; Fernández-Acero, T. Substrates of the MAPK Slt2: Shaping Yeast Cell Integrity. J. Fungi 2022, 8, 368. [Google Scholar] [CrossRef]
- Considine, M.J.; Foyer, C.H. Oxygen and reactive oxygen species-dependent regulation of plant growth and development. Plant Physiol. 2021, 186, 79–92. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Li, R.; Wang, X.; Zhang, S.; Liu, X.; Zhou, Z.; Liu, Z.; Wang, K.; Tian, Y.; Wang, H.; Zhang, Y.; et al. Two zinc-finger proteins control the initiation and elongation of long stalk trichomes in tomato. J. Genet. Genom. 2021, 48, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Orlean, P. Architecture and Biosynthesis of the Saccharomyces cerevisiae Cell Wall. Genetics 2012, 192, 775–818. [Google Scholar] [CrossRef] [Green Version]
- Zan, X.-Y.; Zhu, H.-A.; Jiang, L.-H.; Liang, Y.-Y.; Sun, W.-J.; Tao, T.-L.; Cui, F.-J. The role of Rho1 gene in the cell wall integrity and polysaccharides biosynthesis of the edible mushroom Grifola frondosa. Int. J. Biol. Macromol. 2020, 165, 1593–1603. [Google Scholar] [CrossRef]
- Liu, S.-R.; Zhang, W.-R. Hyperproduction of exopolysaccharides by submerged mycelial culture of Ganoderma lucidum using a solid seed grown in fine-powder of wheat bran and in vitro evaluation of the antioxidant activity of the exopolysaccharides produced. Food Sci. Biotechnol. 2018, 27, 1129–1136. [Google Scholar] [CrossRef]
- Hsieh, C.; Tseng, M.-H.; Liu, C.-J. Production of polysaccharides from Ganoderma lucidum (CCRC 36041) under limitations of nutrients. Enzym. Microb. Technol. 2006, 38, 109–117. [Google Scholar] [CrossRef]
- Levin, D.E. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: The cell wall integrity signaling pathway. Genetics 2011, 189, 1145–1175. [Google Scholar] [CrossRef] [Green Version]
- Orman-Ligeza, B.; Parizot, B.; de Rycke, R.; Fernandez, A.; Himschoot, E.; Van Breusegem, F.; Bennett, M.J.; Périlleux, C.; Beeckman, T.; Draye, X. RBOH-mediated ROS production facilitates lateral root emergence in Arabidopsis. Development 2016, 143, 3328–3339. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Sha, Y.; Wu, D.; Wei, Q.; Chen, D.; Yang, S.; Jia, F.; Yuan, Q.; Han, X.; Wang, J. Surfactant induces ROS-mediated cell membrane permeabilization for the enhancement of mannatide production. Process Biochem. 2020, 91, 172–180. [Google Scholar] [CrossRef]
- Pérez-Pérez, M.E.; Lemaire, S.D.; Crespo, J.L. Control of Autophagy in Chlamydomonas Is Mediated through Redox-Dependent Inactivation of the ATG4 Protease. Plant Physiol. 2016, 172, 2219–2234. [Google Scholar] [CrossRef] [PubMed]
- Ohm, R.A.; de Jong, J.F.; de Bekker, C.; Wösten, H.A.B.; Lugones, L.G. Transcription factor genes of Schizophyllum commune involved in regulation of mushroom formation. Mol. Microbiol. 2011, 81, 1433–1445. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contig | Scaffold | |
|---|---|---|
| Number | 95 | 58 |
| Total length (bp) | 58,157,106 | 58,160,806 |
| N50 (bp) | 2,485,691 | 4,775,195 |
| N90 (bp) | 365,330 | 3,060,068 |
| Maximum length (bp) | 5,247,307 | 8,872,950 |
| GC content (%) | 55.91 | 55.91 |
| Annotated Database | Annotated Number | 100 ≤ length < 300 | Length ≥ 300 |
|---|---|---|---|
| GO_Annotation | 6699 | 1752 | 4827 |
| KEGG_Annotation | 3681 | 1046 | 2561 |
| KOG_Annotation | 6264 | 1509 | 4698 |
| Pfam_Annotation | 8599 | 2102 | 6409 |
| Swissprot_Annotation | 6943 | 1617 | 5248 |
| TrEMBL_Annotation | 13,768 | 3926 | 9646 |
| nr_Annotation | 13,921 | 3999 | 9718 |
| All_Annotated | 13,957 | 4020 | 9733 |
| CAZy_Annotation | 643 | ||
| TCDB_Annotation | 99 | ||
| CYPED_Annotation | 685 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, T.; Cai, M.; Hu, H.; Jiao, C.; Zhang, Z.; Liu, Y.; Chen, J.; Xiao, C.; Li, X.; Gao, X.; et al. Whole-Genome Sequencing and Transcriptome Analysis of Ganoderma lucidum Strain Yw-1-5 Provides New Insights into the Enhanced Effect of Tween80 on Exopolysaccharide Production. J. Fungi 2022, 8, 1081. https://doi.org/10.3390/jof8101081
Wu T, Cai M, Hu H, Jiao C, Zhang Z, Liu Y, Chen J, Xiao C, Li X, Gao X, et al. Whole-Genome Sequencing and Transcriptome Analysis of Ganoderma lucidum Strain Yw-1-5 Provides New Insights into the Enhanced Effect of Tween80 on Exopolysaccharide Production. Journal of Fungi. 2022; 8(10):1081. https://doi.org/10.3390/jof8101081
Chicago/Turabian StyleWu, Tuheng, Manjun Cai, Huiping Hu, Chunwei Jiao, Zhi Zhang, Yuanchao Liu, Jian Chen, Chun Xiao, Xiangmin Li, Xiong Gao, and et al. 2022. "Whole-Genome Sequencing and Transcriptome Analysis of Ganoderma lucidum Strain Yw-1-5 Provides New Insights into the Enhanced Effect of Tween80 on Exopolysaccharide Production" Journal of Fungi 8, no. 10: 1081. https://doi.org/10.3390/jof8101081
APA StyleWu, T., Cai, M., Hu, H., Jiao, C., Zhang, Z., Liu, Y., Chen, J., Xiao, C., Li, X., Gao, X., Chen, S., Wu, Q., & Xie, Y. (2022). Whole-Genome Sequencing and Transcriptome Analysis of Ganoderma lucidum Strain Yw-1-5 Provides New Insights into the Enhanced Effect of Tween80 on Exopolysaccharide Production. Journal of Fungi, 8(10), 1081. https://doi.org/10.3390/jof8101081