Abstract
Transcription factors (TFs) can regulate the synthesis of secondary metabolites through different metabolic pathways in Aureobasidium spp. In this study, a set of 16 superfamilies, 45 PFAM families of TFs with the DNA-binding domains, seven zinc finger families and eight categories of the C2H2 TFs have been identified in Aureobasidium spp. Among all the identified TFs, four superfamilies and six PFAM families are the fungal-specific types in this lineage. The Zn2Cys6 and fungal-specific domain regulators are found to be overwhelmingly predominated, while the C2H2 zinc finger class comprises a smaller regulator class. Since there are currently no databases that allow for easy exploration of the TFs in Aureobasidium spp., based on over 50 references and 2405 homologous TFs, the first TFs pipeline—the Aureobasidium Transcription Factor Database (ATFDB)—has been developed to accelerate the identification of metabolic regulation in various Aureobasidium species. It would be useful to investigate the mechanisms behind the wide adaptability and metabolite diversity of Aureobasidium spp.
1. Introduction
Aureobasidium is a genus of ascomycete in the family Saccotheciaceae that is found worldwide in water, soil, wood, rock, saline habitats, limestone, coastal waters, deep sea, marine sediments from the Antarctic, desert, honeycomb and mangrove ecosystems [1]. This genus is characterized by a high capacity for adaptation to a complex and changing living environment. Diverse strains of Aureobasidium spp. produce a variety of secondary metabolites, including polymalate (PMA), pullulan, liamocins, siderophores, melanin, polyunsaturated fatty acids (PUFA), massoia lactone, intracellular lipids, gluconic acids (GA), fructooligosaccharides (FOSs) and various enzymes, indicating that they have potential applications in biotechnology [2,3,4,5,6]. The biosynthesis of secondary metabolites in Aureobasidium spp. is intimately related to the distinct signal pathways, the whereabouts of carbon metabolic flow and regulation of various transcription factors (TFs) [7,8]. Transcription factors are recognized to have a vital role in influencing the levels of pathway gene expression, hence regulating flow via secondary metabolic pathways [9,10].
Until now, numerous different types of transcription factors have been intensively studied in yeast, including Aureobasidium spp. and the related Aspergillus species for their functions and regulatory mechanisms. [9,10]. Previously published research studies have established that the types of TFs in Aureobasidium spp. were much lower than those in Aspergillus spp., Saccharomyces cerevisiae, and other eukaryotes in the TRANSFAC (http://gene-regulation.com/) (accessed on 20 January 2019) and the YEASTRACT+ (http://www.yeastract.com) databases [10,11,12,13,14,15]. However, due to the high genomic diversity of Aureobasidium spp., representative data in current databases are underrepresented. Especially, many transcription factors lack the KEGG Orthologs (KO) (https://www.kegg.jp/kegg/ko.html, accessed on 14 April 2022). Meanwhile, no publications on the detailed classification, regulatory mechanism, or function of TFs in the Aureobasidium genus have been published, and no tools exist to effectively integrate current biological knowledge or transcription regulation of Aureobasidium spp. with the keywords of TF terms submitted by users. This hinders the study of the transcription regulation mechanisms of this genus.
This study manually selected and categorized the TF families and constructed a complete gene set—the “Aureobasidium Transcription Factor Database (ATFDB)” to retrieve TFs and download the Hidden Markov Models (HMMs) based on user-supplied search keywords. In order to widely search orthologs against this database, each type of the TFs is trained into the HMM-based profile. Users can quickly retrieve information by entering the names of different transcription factors or the types of TFs. For example, if you type Msn2, you obtain the relevant information about the global transcription factor Msn2 in Aureobasidium spp., and if you type C2H2, you obtain a TFs list of all these types in Aureobasidium spp. Not only does the ATFDB provide a minimum gene set for TFs in the Aureobasidium genus, but it also has thorough detailed information on TFs sharing among different strains of Aureobasidium spp. We have established an integrated TFs repertoire in the Aureobasidium spp., which promotes the discovery of transcription regulation in distinct Aureobasidium species.
2. Methods
2.1. Genome Collection and Data Analysis
A total of 1,578,889 protein sequences from 146 strains of Aureobasidium spp. are retrieved and manually downloaded from the GeneBank database (https://www.ncbi.nlm.nih.gov/assembly/) (accessed on 27 March 2022). Additionally, the representative genomes of the pullulan-producing strain Aureobasidium melanogenum P16, four reference strains of Aureobasidium spp. and fifty strains of Aureobasidium pullulans from various sources have been categorized for further investigation (Table S1) [16,17].
2.2. Homologues TFs Searches and Training
The TF genes from the genomes of S. cerevisiae and Aspergillus spp. are utilized as a reference for searching the publicly available, assembled genomes of the Aureobasidium strains in the NCBI database (https://www.ncbi.nlm.nih.gov/). To identify the TFs, we used BLAST 2.5.0+ and BioEdit version 7.0.9.0 to search for homologous TFs in the genomes of Aureobasidium spp. The Hidden Markov Models (HMM) of various TFs are trained through the HMMER 3.3.2 software by using default parameters. The PFAM families and super-families of the DNA-binding domains are estimated and sorted through the Pfam (http://www.pfam.org/) and the SUPERFAMILY 2.0 web server (http://supfam.org).
2.3. Function Annotation and Domain Analysis
All the TFs are annotated in the GhostKOALA, the Cluster of Orthologous Groups of proteins (COG), Pfam, the Non-Redundant Protein Sequence Database (NR) and the Swiss-Prot database as previously described [18,19,20]. Each of them is summarized in reference to the YEASTRACT+ database and the published references for inferring the functions. The domains of the TFs are exhibited through the TBtools v1.098652 software.
2.4. Compilation of the TF Genes
All of the TFs’ information and sequences are publicly available on the ATFDB, which may be searched using keywords. The TF names are directly utilized as inputs (multiple inputs available) in the ATFDB, which can be used to search for their features and accession numbers. There are 45 different types of TFs that can be obtained, each with all of their feature annotations applied manually (Table S2). Furthermore, we make every attempt to ensure that gene sequences for each kind of TF are obtained and downloaded for additional research.
2.5. TFs Classification between the NCBI and Transcription Factor Database
The TF classification assignments in the ATFDB and NCBI databases are compared throughout the genomes of Aureobasidium spp. in the GenBank database [21,22]. The classification of various transcription factors and the involved metabolic pathways is conducted based on the public articles. The sankey diagram is built in R 3.0.1+ by using the networkD3 package (https://www.r-graph-gallery.com/).
2.6. Database Construction
We aimed to construct ATFDB for comprehensive, accurate and rapid analysis of the transcriptional regulation mechanisms in the environmental adaptability of Aureobasidium spp. Thus, the constructed database should contain the most comprehensive and precise TFs gene families based on the current knowledge. In addition, TFs and their homologues from multiple orthology databases should be included to reduce false-positive assignments. Therefore, a pipeline was developed for ATFDB and included the following steps (Figure 1).
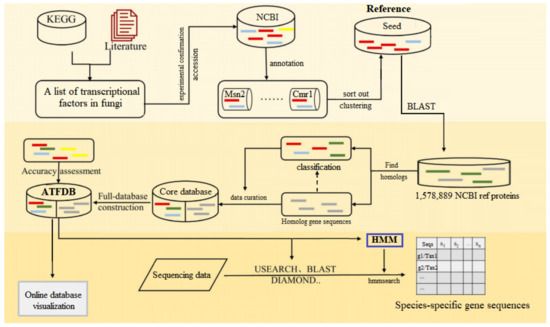
Figure 1.
Flowchart of major steps of ATFDB construction. First, a seed database was constructed for selected genes by retrieving protein sequences from the NCBI database using the accession numbers. Second, target genes from the NCBI and their homologs were identified and integrated to construct the core database. At last, all data were uploaded to the online database for visualization, and the trained HMM models were developed to conduct species-specific TFs profiling of Aureobasidium spp. via the sequencing data.
3. Results and Discussion
3.1. PFAM Families of DNA-Binding Domains in Aureobasidium spp.
The PFAM database currently has 670 entries for the term “DNA binding” (Interpro: 1053) of the PFAM families. After text-mining-based filtering of the general TFs, the putative and basal metabolic proteins, 45 common PFAM families were finally identified and selected for analysis based on these family entries (Table 1). Additionally, 16 superfamilies were predicted among the total number of superfamilies of DNA-binding domains in reference to the type strain Aureobasidium melanogenum CBS110374. Among all these 16 superfamilies, 4 superfamilies were classified as specific transcription factors: the Zn2/Cys6 (Zn cluster), the DNA-binding domain of Mlu1-box-binding protein Mbp1, the basic-leucine zipper (bZIP) and the Zinc domain/Copper fist DNA binding domain (Table 1).

Table 1.
Sixteen superfamilies and forty-five PFAM families of TF DNA binding domains predicted to occur in Aureobasidium spp. (except for general transcription factors, DNA repair/extend, nuclear receptor and chromatin remodeling).
3.2. The Zn2Cys6 and Fungal-Specific TF Families
Four superfamilies and six PFAM families of the TFs were identified as the fungal-specific TFs in Aureobasidium spp. (Table 2). In fungi, these TFs are genus-specific and quite function-specific, regulating the fungal-specific morphogenetic processes that remain ubiquitous and conservative functions in Aureobasidium spp. The analysis results of the 45 fungal transcription regulator-related PFAM domains suggest that there are significant differences in the number of transcription regulators across different fungal classes (Figure 2 and Figure S1). The fungal specific transcription factor domain (PF04082) and the fungal Zn(2)-Cys(6) binuclear cluster domain (PF00172) comprise a large proportion in different fungi (Figure 2). The numbers of the two Pfam families were found to be larger in Aspergillus spp. and Aureobasidium spp. than that in S. cerevisiae (Figure S1). These findings indicate that since the Aureobasidium lineage separated, different classes of fungi have evolved different regulatory mechanisms.
Table 2.
The Zn2Cys6 and fungal-specific TFs in Aureobasidium spp.
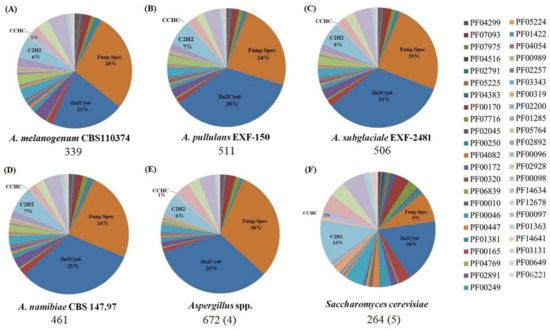
Figure 2.
Distribution of PFAM families in different fungi. (A) A. melanogenum CBS110374; (B) A. pullulans EXF-150; (C) A. subglaciale EXF-2481; (D) A. namibiae CBS 147.97; (E) Aspergillus spp.: A. aculeatus, A. niger, A. fumigatis and A. oryzae RIB40; (F) S. cerevisiae: S. cerevisiae S288C, S. cerevisiae YJM244, S. cerevisiae YJM450, S. cerevisiae YJM993 and S. cerevisiae YJM1078. The average number of the regulators for each species is indicated underneath each pie chart. The number of genomes analyzed in all species is indicated in parentheses.
In S. cerevisiae, the total number of the regulators is very low, which is consistent with the general PFAM distribution. Generally, the Zn2Cys6 and fungal-specific domain regulators have overwhelmingly predominated in Aureobasidium spp., whereas the C2H2 zinc finger class is a much lesser regulator class. However, compared with that in S. cerevisiae, the proportion of the C2H2 types decreased significantly in Aureobasidium spp. and Aspergillus spp. (Figure 2). Furthermore, the classification, function, and regulation of these fungal-specific TF families have been further elucidated as described below, including zinc cluster TF families, fungal-specific domain TF families, KilA-N domain TF families, HMG-box domain TF families, basic Leu zipper (bZIP) families and forkhead TF families.
3.2.1. The Zinc Cluster TF Families
The Zn2/Cys6 (Zn cluster) superfamily (PF00172) is a vast group of the fungal-specific TFs that regulate many critical metabolic processes involved in cell growth and development [15]. Likewise, the Zn(II)2Cys6 domain family proteins in Aureobasidium spp. (numbers: 141) and Aspergillus (numbers: 222) are greater than that in S. cerevisiae (numbers: 48) (Figure 2), suggesting that the major expansion of these regulator classes may have occurred following the divergence of Aureobasidium spp. and Aspergillus spp. at ancient times [18]. For tuning iron uptake and storage of Aureobasidium melanogenum HN6.2, the transcriptome analysis demonstrates that the zinc cluster transcription factor SreA regulates and maintains iron homeostasis (Table S2) [23].
3.2.2. The “Fungal-Specific Transcription Factor Domain” TF Families
The second large class of TFs is the “fungal-specific transcription factor domain” (PF04082, IPR007219), which has expanded more rapidly in the Ascomycota than in the Basidiomycota [24]. In Aureobasidium spp., it consists of three members: the Fungal trans (PF04082), the Cep3 (PF16846), and the Fungal trans 2 (PF04082 or PF11951). Transcription factors with this domain are involved in the metabolic flow of carbon pool and fatty acids (Table 2).
3.2.3. The KilA-N Domain TF Families
The KilA-N domain (PF04383) has been verified to regulate cellular differentiation, melanin synthesis, cell growth, and development in Aureobasidium spp. Meanwhile, ankyrins are multifunctional adaptors that link specific proteins to the membrane-associated, spectrin-actin cytoskeleton. The results show that although there are no differences in the KilA-N domain of Swi4/6 among different fungi, in Aureobasidium spp., transcription factor Swi4/6 has a long sequence coverage of the ankyrin repeats (ANK) with at least three consecutive copies, which is obviously different from those in A. fumigatus and S. cerevisiae (Figure 3) [25].
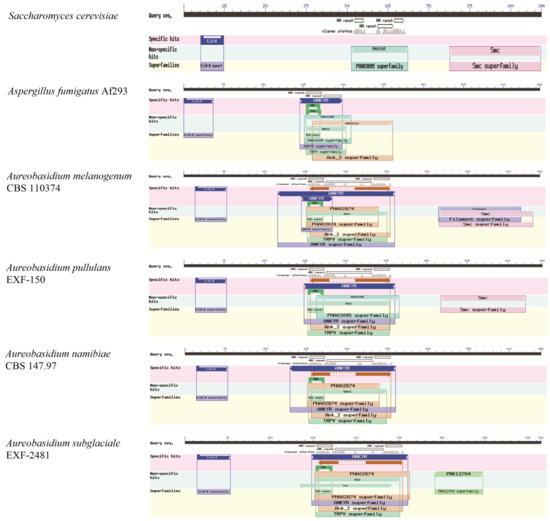
Figure 3.
Conserved functional domains of the KilA-N type protein Swi4.
3.2.4. The HMG-Box Domain TF Families
3.2.5. The HTH, Basic Leu Zipper (bZip) and Forkhead (FH) TF Families
The HTH, the GATA zinc finger, and the basic Leu zipper motif (bZIP) are also classified as yeast-specific TFs [26]. The βHLH regulator involves the homologue of Ino4 (KEQ67287.1) in Aureobasidium spp., which is equivalent to the regulator of phospholipid synthesis in S. cerevisiae. Sre1 (KEQ62711.1) and SreA (KEQ65560.1) are identified in the genome of Aureobasidium spp., of which they are transcription repressors that inhibit the biosynthesis of iron carriers in Aureobasidium spp. The bZIP proteins mainly are primarily involved with the iron acquisition regulator HapX in the synthesis of siderophore (Table S2).
3.3. Identification of Transcription Factors in Aureobasidium spp.
The transcription factors of Aureobasidium spp. are identified by searching annotated genes and comprehensive results of biochemical and genetic analysis of S. cerevisiae and Aspergillus fumigatus. Generally, the homologous TFs found in Aureobasidium spp. are manually curated according to the literature of previous research (Table S2). Forty-five transcription factors characterize and represent a variety of distinct functions in Aureobasidium spp., including six involved in the nitrogen metabolism (Seb1/Msn2, AreA, AreB, MeaB, TamA and Put3), five involved in synthesis of siderophores (AcuM, Sre1, SreA, HapX and Hap2), four involved in synthesis of polysaccharides (XlnR/GliZ, Gal4, AmyR and Ftr1), two involved in the glycolysis (Gcr1), and gluconeogenesis regulation (Cat8), while six general TFs are associated with RNA polymerase II transcription (TFⅡA, TFⅡB, TFⅡD, TFⅡE, TFⅡF and TFⅡH), and another six are involved in the signal pathway regulation, such as the HOG, cAMP-PKA, MAPK, and CWI pathways, as well as stress response pathway (Table S2). Approximately three metabolic pathway-specific transcription factors are found to be associated with the regulation of secondary metabolites, such as the homologues transcriptional activator AflR, Cmr1/Pig1 and the GATA-type sexual development transcription factor NsdD (Table S2).
Additionally, the other regulators mainly participate in the biochemical processes of phospholipid and fatty acid synthesis, chromatin remodeling, DNA repair, oxidative stress response, heat shock response, and protein folding (Table S2). The transcriptional activator Hsf1 (KEQ66690.1), for example, can specifically recognize the nGAAn repeat units in the promoter region of heat shock elements (HSEs), therefore regulating heat shock proteins for thermotolerance [27]. It is widely distributed in the genomes of Aureobasidium spp. (Table S2). However, most of the homologues involved in the carbon metabolism (GalX, GalR, ScfA, AlcR, AceII) are found to be lost in Aureobasidium spp., indicating a genus specificity in Aspergillus spp.
3.3.1. Taxonomy and Functions of the Zinc Finger TFs in Aureobasidium spp.
The Zinc finger (PFAM entries: 257) exists in various proteins and has a wide range of functions in various cellular processes [28]. There are 215 genes encoding zinc finger proteins in the genomes of Aureobasidium spp., the number of which is comparable to that of S. cerevisiae (217) and Aspergillus niger (2951). Initially, zinc fingers were categorized according to the different numbers and orders of the residues (Cys2His2, Cys4, and Cys6). Krishna et al. [28] previously described a more systematic approach for classifying zinc finger proteins into “fold groups” based on the overall structure of the protein backbone. The PFAM database presently contains 257 zinc finger families; thus, after manually filtering the unrelated and unknown functional proteins, we finally summarized the identified zinc finger families into seven “fold groups” in Aureobasidium spp. as follows (Table S3). Seven types of zinc finger transcription factors (class Ⅰ-Ⅶ) have been found in the genomes of Aureobasidium spp. (Table S3), which are widely involved in the regulation of osmotic pressure regulation, secondary metabolites, the MAPK pathway, the calcium signaling pathway and glucose suppression (Table S2).
3.3.2. Classification and Functions of the C2H2 TFs in Aureobasidium spp.
The C2H2 and C2HC motifs are highly conserved in the majority of fungi, and they are involved in transcription regulation [9]. We searched for the C2H2 TFs in the genomes of Aspergillus aculeatus ATCC16872, A. melanogenum P16, A. melanogenum CBS110374, Aureobasidium subglaciale EXF-2481, Aureobasidium pullulans EXF-150 and Aureobasidium namibiae CBS147.97, and the neighbor-joining (NJ) phylogenies for the datasets of sequences were then inferred by MEGA v7.0.26. According to the different domains and functions of the C2H2 TFs, the homologues can be divided into eight categories in different strains of Aureobasidium spp. (Figure 4A), which mainly contain the global transcription factors, specifically TFs, and metabolic pathways of specific transcription factors (Table S2).
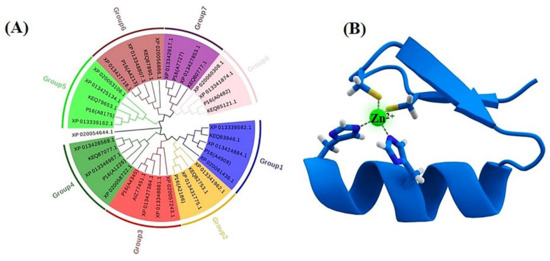
Figure 4.
Phylogenetic tree of the C2H2 transcription factors (A) and the Cys2His2 zinc finger motif, consisting of α helix and an antiparallel β sheet (B). The zinc ion (green) is coordinated by two histidine residues and two cysteine residues.
Global transcription factors are classified into four groups (groups 1, 4, 5 and 6), such as the transcription repressor CreA/Mig1 (KEQ62800.1), the transcriptional activator Seb1/Msn2 (KEQ58292.1), Crz1 (KEQ62890.1) and Ste12/BrlA (KEQ57897.1) (Figure 4A). In Aureobasidium spp., CreA is known to be important in the regulation of pullulan biosynthesis, and deletion of the CREA/MIG1 gene causes de-repression of the expression of several genes in A. melanognum P16 under high-concentration glucose medium [29]. Seb1/Msn2, a global transcription factor, acts as a regulator in the osmotic pressure response pathway to glycogen accumulation. The cellular localization of Msn2 is determined by phosphorylation, and the dephosphorylated Msn2 is clustered in the cell nuclei to activate the expression of the UGP1 gene and regulates pullulan production in A. melanogenum P16 [7,30]. Crz1 commonly regulates gene transcription in response to environmental changes through dephosphorylation by a Ca2+/calmodulin-dependent phosphatase [31]. PMA biosynthesis is regulated by the transcriptional activator Crz1 from the Ca2+ signaling pathway [32,33], and this transcriptional activator can be found in most Aureobasidium genomes (Table S2). The global transcription factor Ste12 and Ste12-like proteins (KEQ57897.1) are found exclusively in fungi that are activated by a cascade of mitogen-activated protein kinase (MAPK) signals. The C-terminal C2H2-Zn2+ finger domains are found in Aureobasidium spp. and Aspergillus spp. but not in S. cerevisiae, Yarrowia lipolytica, Kluyveromyces marxianus and Candida albicans [32,33]. Fungal-specific transcription factors contain two groups (groups 2 and 3), such as the PacC (KEQ63386.1) and Mac1 (KEQ60235.1) (Figure 4A). PacC is activated by a signal transduction pathway triggered by neutral or alkaline pH [34]. Another one, the fungal-specific transcription factor Mac1, is indispensable to switches of gene expression required for high affinity copper ion transport [35,36]. Metabolic pathway-specific transcription factors are also classified into two groups (groups 7 and 8), including ScpR/AfoA (KEQ63975.1) and Cmr1/Pig1 (KEQ67497.1) (Figure 4A) [25,37]. There are special amino acid residues of DNA interaction—R, H, R and R, E, R in the C2H2 Zn finger of Aureobasidium spp. (Figure 4B). ScpR/AfoA can regulate the aspartate gene cluster, NRPS gene (inpA and inpB) and biosynthesis of asper furanone expression [35]. In fungi, the PKS1 gene and CMR1 gene links together and constitutes a gene cluster to catalyze melanin synthesis. The metabolic pathway-specific transcription factor Cmr1 can regulate the expression of the PKS gene in Aureobasidium spp. [25]. Cmr1 has three types of functional motifs as mentioned above (Figure 5), indicating that it is a unique metabolic pathway specific transcription factor. It has similar domains in the proximal species and may only exist in most asexual lineages of Dothideomycetes, such as Aureobasidium spp. and other melanin-producing fungi (Figure 5). In contrast, it was found to be lost in the Aspergillus of Ascomycete and is only confined to part of the melanin-producing fungi, indicating that Cmr1 is also a species-specific transcription factor [25] (Figure 5).

Figure 5.
Transcription factor Cmr1 and the target protein Pks1 in different fungi.
3.4. Transcription Factor Database for Aureobasidium spp.
To establish a complete gene set of TFs in the Aureobasidium genus, we investigated the reported transcription factors in the published genomes of Aureobasidium spp. Finally, we identified a total of 5494 common homologous TFs in 1,578,889 proteins from 146 strains of Aureobasidium spp. using the reference strain A. melanogenum CBS110374. The identity thresholds and gene accessions can be checked in Table S4. Further, the intact Hidden Markov Models (HMM) of different TFs have been perfectly trained and uploaded to the Aureobasidium Transcription Factor Database, which can be utilized to find more orthologous or paralogous TFs in this genus (see the HMMER 3.3.2 usage). This is more accurate than directly using the type strain for sequence alignment, because the genomes of type strains such as A. melanogenum CBS110374 or A. pullulans EXF-150 are the draft genomes with many genes to be incompletely assembled. In addition, the TFs in each of the fifty A. pullulans strains are chosen as the representative gene set for analysis since A. pullulans is the common species of Aureobasidium [16,17].
After submitting all the data, the Aureobasidium Transcription Factor Database has been established for TF retrieval. The database involves four sections and works on an intuitive approach: (i) users input the retrieval data; (ii) automatic word matching; (iii) query by exactly matching the database; (ⅳ) process and encapsulate the retrieved data format; (ⅴ) return the encapsulated data and render the returned data to show the results (Figure 6). All the data in the ATFDB is publicly accessible at https://huang.zgsj1.com/picture/literature/distweb/index.html (accessed on 15 April 2022), which also supports multiple inputs for users to retrieve. Meanwhile, all of the TFs can be clicked for information linking to external databases, and the KEGG entries for TFs can also be checked in the KEGG section. The user-friendly web server interface accepts user input and produces results in seconds.
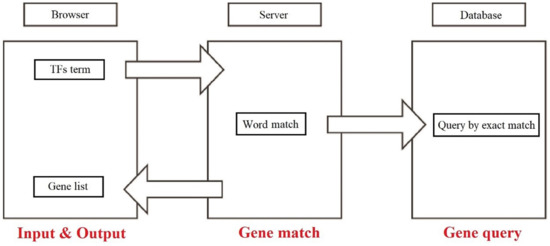
Figure 6.
Workflow of the Aureobasidium Transcription Factor Database.
We tested the ATFDB on a small dataset of the known C2H2 TFs. For example, when users search for the term “Crz1”, the detailed contents of the message are shown, including the transcription factors’ accession numbers and information relating to other databases. Furthermore, when entering the term “C2H2”, users can quickly retrieve the common transcription factors of this type, such as the transcription activator Cmr1 and the transcription repressor Mig1/CreA (Figure S2). This may also be used to search for various TFs in Aureobasidium spp., such as the Zn2Cys6, bZIP and GATA zinc fingers, demonstrating the database’s comprehensiveness and usefulness (Figure S2). Additionally, we provided the representative protein sequences of transcription factors from each of the fifty A. pullulans strains for users to download for further study. The ATFDB exhibits the known transcription regulation and gene associations of several TFs, highlighting its ability to contribute to the formulation of novel biological hypotheses. It is capable of identifying a significant number of candidate transcriptional regulators in Aureobasidium spp., many of which have been verified and studied in experiments [2,3,7,13,14,38,39,40,41]. Due to the increasing number of genome sequencing studies, no other approach, including YEASTRACT+, the NCBI, or the TRANSFAC database, could be used to obtain the species-specific TFs of Aureobasidium spp. The ATFDB can meet the needs of users by leveraging information about prior biological knowledge and phenotype to expedite and announce scientific discoveries.
3.5. Advantages of the ATFDB over Other Orthology Databases
In order to compare the differences between the NCBI and the ATFDB databases, we searched in NCBI through keywords according to different types of transcription factors as described above, such as “Msn2 Aureobasidium”. We then classified them according to the categories as stated above (including global, general and specific TFs) and counted the number of TFs contained within different categories. The results showed that in the type-level of TF classification, the NCBI (GenBank) database mostly contained general TFs (2511), but the ATFDB largely compensated for the NCBI (GenBank) database’s faults and weaknesses in the global TFs (1386) and particular TFs (697) (Figure 7A) (Table S5). In particular, many TFs in the NCBI database are termed as hypothetical or putative proteins and are not further classified, which is inconvenient for the study of gene diversities. Apart from the fundamental metabolisms, the ATFDB outperforms the NCBI (GenBank) database in the signal pathways (426), secondary metabolisms (589), and certain other metabolic reaction processes (369) (Figure 7B) (Table S5), suggesting that it complements and covers the majority of the common transcription factors. The classification of TFs and their participating metabolic pathways in the ATFDB are evaluated by comparison with that of the NCBI (GenBank) database.
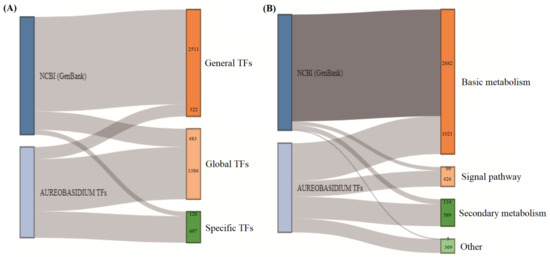
Figure 7.
Comparison of the TFs numbers of Aureobasidium spp. in the NCBI (GenBank) and the ATFDB databases. (A) Comparison of the NCBI (GenBank) and the ATFDB databases at type-level of TFs classifications. (B) Comparison of the NCBI (GenBank) and the ATFDB databases at the metabolism-level of TFs classifications.
4. Conclusions
The lineage of Aureobasidium spp. can synthesize a variety of natural compounds with high-economy value and potential industrial applications, the majority of which are associated with transcription factor metabolic regulatory networks. The built ATFDB can be applied for multiple purposes. Genes involved in each pathway are comprehensively and accurately presented in ATFDB. First, it holds the potential to be further used for genomic annotation. For example, if all transcriptional regulations involved in one pathway are annotated, we can consider the mechanisms of metabolism potentials of this pathway. In addition, ATFDB may be used for analyzing amplicon sequencing data of functional genes to separate homologous groups. In summary, this study presents a manually constructed database (ATFDB) that aims to profile transcription factor metabolic regulatory networks. It is publicly available without login requirements at https://huang.zgsj1.com/picture/literature/distweb/index.html (accessed on 14 April 2022). The ATFDB contains 16 superfamilies, 45 PFAM families of TFs with DNA-binding domains, 7 zinc finger families, and 8 categories of the C2H2-type TFs. The results demonstrate that ATFDB is a useful tool for studying the transcriptional regulations of Aureobasidium spp. in the environment, and it will be continuously updated.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jof8101096/s1, Figure S1: The numbers of the PFAM families in different fungi; Figure S2: The retrieval and KEGG annotation interface of Crz1 or C2H2 in Aureobasidium Transcription Factor Database; Table S1:Accession numbers of the Aureobsidium genome sequences included in this study; Table S2: Putative transcription factor involved in regulating the response to osmotic stress; member of the MADS-box family of transcription factors; Table S3: Major zinc finger transcriptional factors in Aureobasidium spp.; Table S4: A total of 5,494 common homologous TFs in 1,578,889 proteins from 146 strains of Aureobasidium spp. using the reference strain A. melanogenum CBS110374.; Table S5: The TFs numbers of Aureobasidium spp. in the NCBI (GenBank) and the ATFDB databases.
Author Contributions
Conceptualization, Y.F.; methodology, Z.H., and S.J.; software, Z.H. and Z.C.; formal analysis, H.M. and S.L.; investigation, H.M. and Z.C.; resources, X.H.; data curation, G.Y.; writing—original draft preparation, G.Y., Y.F., Y.W. and S.J.; writing—review and editing, G.Y., Y.W. and S.J.; project administration, X.H.; funding acquisition, G.Y. and X.H. All authors have read and agreed to the published version of the manuscript.”
Funding
This work was supported by the National Natural Science Foundation of China (32102270), the National Natural Science Foundation of Jiangsu Ocean University (KQ20041), Project “333” of Jiangsu Province, Open-end Funds of Jiangsu Institute of Marine Resources Development (JSIMR202024) and Open-end Funds of Jiangsu Key Laboratory of Marine Bioresources and Environment (SH20211210).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data are incorporated into the article and the Aureobasidium Transcription Factor Database (https://huang.zgsj1.com/picture/literature/distweb/index.html, accessed on 15 April 2022). The data underlying this article are available in the article and in its online supplementary material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jia, S.L.; Chi, Z.; Liu, G.L.; Hu, Z.; Chi, Z.M. Fungi in mangrove ecosystems and their potential applications. Crit. Rev. Biotechnol. 2020, 40, 852–864. [Google Scholar] [CrossRef]
- Zhang, M.; Gao, Z.C.; Chi, Z.; Liu, G.L.; Hu, Z.; Chi, Z.M. cAMP-PKA and HOG1 signaling pathways regulate liamocin production by different ways via the transcriptional activator Msn2 in Aureobasidium melanogenum. Enzyme Microb. Technol. 2021, 143, 109705. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chi, Z.; Liu, G.L.; Qi, C.Y.; Jiang, H.; Hu, Z.; Chi, Z.M. A novel PMA synthetase is the key enzyme for polymalate biosynthesis and its gene is regulated by a calcium signaling pathway in Aureobasidium melanogenum ATCC62921. Int. J. Biol. Macromol. 2020, 156, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.J.; Chen, L.; Jiang, H.; Liu, G.L.; Chi, Z.M.; Hu, Z.; Chi, Z. High pullulan biosynthesis from high concentration of glucose by a hyperosmotic resistant, yeast-like fungal strain isolated from a natural comb-honey. Food Chem. 2019, 286, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Danesi, P.; James, T.Y.; Al-Hatmi, A.; Najafzadeh, M.J.; Dolatabadi, S.; Ming, C.; Liou, G.Y.; Kang, Y.; de Hoog, S. Comparative pathogenicity of opportunistic black yeasts in Aureobasidium. Mycoses 2019, 62, 803–811. [Google Scholar] [PubMed]
- Aung, T.; Jiang, H.; Liu, G.L.; Chi, Z.; Hu, Z.; Chi, Z.M. Overproduction of a beta-fructofuranosidase1 with a high FOS synthesis activity for efficient biosynthesis of fructooligosaccharides. Int. J. Biol. Macromol. 2019, 130, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Liu, G.L.; Wang, S.J.; Chi, Z.M.; Chi, Z. Pullulan biosynthesis in yeast-like fungal cells is regulated by the transcriptional activator Msn2 and cAMP-PKA signaling pathway. Int. J. Biol. Macromol. 2020, 157, 591–603. [Google Scholar] [CrossRef]
- Slj, A.; Zhe, C.; Lu, C.; Glla, B.; Zhong, H.C.; Zmca, B. Molecular evolution and regulation of DHN melanin-related gene clusters are closely related to adaptation of different melanin-producing fungi. Genomics 2021, 113, 1962–1975. [Google Scholar]
- Shelest, E. Transcription factors in fungi. FEMS Microbiol. Lett. 2008, 286, 145–151. [Google Scholar] [CrossRef]
- Drobna, E.; Bialkova, A.; Subik, J. Transcriptional regulators of seven yeast species: Comparative genome analysis. Review. Folia Microbiol. 2008, 53, 275–287. [Google Scholar] [CrossRef]
- Shelest, E. Transcription factors in fungi: Tfome dynamics, three major families, and dual-specificity TFs. Front. Genet. 2017, 8, 53. [Google Scholar] [CrossRef]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.; Otillar, R.; Riley, R.; Salamov, A.; Zhao, X.; Korzeniewski, F.; et al. Mycocosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014, 42, D699–D704. [Google Scholar] [CrossRef]
- Kang, X.X.; Jia, S.L.; Wei, X.; Zhang, M.; Liu, G.L.; Hu, Z.; Chi, Z.; Chi, Z.M. Liamocins biosynthesis, its regulation in Aureobasidium spp., and their bioactivities. Crit. Rev. Biotechnol. 2022, 42, 93–105. [Google Scholar] [CrossRef]
- Wei, X.; Liu, G.L.; Jia, S.L.; Chi, Z.; Hu, Z.; Chi, Z.M. Pullulan biosynthesis and its regulation in Aureobasidium spp. Carbohydr. Polym. 2021, 251, 117076. [Google Scholar] [CrossRef]
- Macpherson, S.; Larochelle, M.; Turcotte, B. A fungal family of transcriptional regulators: The zinc cluster proteins. Microbiol. Mol. Biol. R. 2006, 70, 583–604. [Google Scholar] [CrossRef]
- Gostincar, C.; Turk, M.; Zajc, J.; Gunde-Cimerman, N. Fifty Aureobasidium pullulans genomes reveal a recombining polyextremotolerant generalist. Environ. Microbiol. 2019, 21, 3638–3652. [Google Scholar] [CrossRef]
- Gostincar, C.; Ohm, R.A.; Kogej, T.; Sonjak, S.; Turk, M.; Zajc, J.; Zalar, P.; Grube, M.; Sun, H.; Han, J.; et al. Genome sequencing of four Aureobasidium pullulans varieties: Biotechnological potential, stress tolerance, and description of new species. BMC Genom. 2014, 15, 549. [Google Scholar] [CrossRef]
- Jia, S.L.; Ma, Y.; Chi, Z.; Liu, G.L.; Chi, Z.M. Genome sequencing of a yeast-like fungal strain P6, a novel species of Aureobasidium spp.: Insights into its taxonomy, evolution, and biotechnological potentials. Ann. Microbiol. 2019, 69, 1475–1488. [Google Scholar] [CrossRef]
- Yang, G.; Cui, X.; Liu, S.; Lu, J.; Fang, Y. Effects of dietary Lactobacillus helveticus on the growth rate, disease resistance and intestinal health of pond loach (Misgurnus anguillicaudatus). Aquaculture 2021, 544, 737038. [Google Scholar] [CrossRef]
- Tsang, L.M.; Ming, L.; Shen, X.; Cheang, C.C.; Chu, K.H.; Chan, B.K.K. Gene rearrangement and sequence analysis of mitogenomes suggest polyphyly of archaeobalanid and balanid barnacles (Cirripedia: Balanomorpha). Zool. Scr. 2017, 46, 729–739. [Google Scholar] [CrossRef]
- Liu, F. Chloroplast genome of Sargassum horneri (Sargassaceae, Phaeophyceae): Comparative chloroplast genomics of brown algae. J. Appl. Phycol. 2016, 28, 1419–1426. [Google Scholar] [CrossRef]
- Shen, X.; Ling, M.T.; Chu, K.H.; Achituv, Y.; Chan, B. Mitochondrial genome of the intertidal acorn barnacle Tetraclita serrata Darwin, 1854 (Crustacea: Sessilia): Gene order comparison and phylogenetic consideration within Sessilia. Mar. Genom. 2015, 22, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, G.; Jiang, H.; Chi, Z.; Chi, Z. An insight into the iron acquisition and homeostasis in Aureobasidium melanogenum HN6.2 strain through genome mining and transcriptome analysis. Funct. Integr. Genom. 2019, 19, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.B.; Zhou, M.; Ohm, R.A.; Leeggangers, H.A.; Visser, L.; de Vries, R.P. Prevalence of transcription factors in ascomycete and basidiomycete fungi. BMC Genom. 2014, 15, 214. [Google Scholar] [CrossRef]
- Jiang, H.; Chi, Z.; Liu, G.L.; Hu, Z.; Zhao, S.Z.; Chi, Z.M. Melanin biosynthesis in the desert-derived Aureobasidium melanogenum XJ5-1 is controlled mainly by the CWI signal pathway via a transcriptional activator Cmr1. Curr. Genet. 2020, 66, 173–185. [Google Scholar] [CrossRef]
- Amoutzias, G.D.; Veron, A.S.; Weiner, J.R.; Robinson-Rechavi, M.; Bornberg-Bauer, E.; Oliver, S.G.; Robertson, D.L. One billion years of bZIP transcription factor evolution: Conservation and change in dimerization and DNA-binding site specificity. Mol. Biol. Evol. 2007, 24, 827–835. [Google Scholar] [CrossRef]
- Hahn, J.S.; Hu, Z.; Thiele, D.J.; Iyer, V.R. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell. Biol. 2004, 24, 5249–5256. [Google Scholar] [CrossRef]
- Krishna, S.S.; Majumdar, I.; Grishin, N.V. Structural classification of zinc fingers: Survey and summary. Nucleic Acids Res. 2003, 31, 532–550. [Google Scholar] [CrossRef]
- Wang, Q.Q.; Lu, Y.; Ren, Z.Y.; Chi, Z.; Liu, G.L.; Chi, Z.M. CreA is directly involved in pullulan biosynthesis and regulation of Aureobasidium melanogenum P16. Curr. Genet. 2017, 63, 471–485. [Google Scholar] [CrossRef]
- Gorner, W.; Durchschlag, E.; Martinez-Pastor, M.T.; Estruch, F.; Ammerer, G.; Hamilton, B.; Ruis, H.; Schuller, C. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 1998, 12, 586–597. [Google Scholar] [CrossRef]
- Stathopoulos-Gerontides, A.; Guo, J.J.; Cyert, M.S. Yeast calcineurin regulates nuclear localization of the Crz1p transcription factor through dephosphorylation. Genes Dev. 1999, 13, 798. [Google Scholar] [CrossRef]
- van der Felden, J.; Weisser, S.; Bruckner, S.; Lenz, P.; Mosch, H.U. The transcription factors Tec1 and Ste12 interact with coregulators Msa1 and Msa2 to activate adhesion and multicellular development. Mol. Cell. Biol. 2014, 34, 2283–2293. [Google Scholar] [CrossRef]
- Borneman, A.R.; Hynes, M.J.; Andrianopoulos, A. An STE12 homolog from the asexual, dimorphic fungus Penicillium marneffei complements the defect in sexual development of an Aspergillus nidulans steA mutant. Genetics 2001, 157, 1003–1014. [Google Scholar] [CrossRef]
- Barda, O.; Maor, U.; Sadhasivam, S.; Bi, Y.; Zakin, V.; Prusky, D.; Sionov, E. The pH-responsive transcription factor PacC governs pathogenicity and ochratoxin a biosynthesis in Aspergillus carbonarius. Front. Microbiol. 2020, 11, 210. [Google Scholar] [CrossRef]
- Chiang, Y.M.; Szewczyk, E.; Davidson, A.D.; Keller, N.; Oakley, B.R.; Wang, C.C. A gene cluster containing two fungal polyketide synthases encodes the biosynthetic pathway for a polyketide, asperfuranone, in Aspergillus nidulans. J. Am. Chem. Soc. 2009, 131, 2965–2970. [Google Scholar] [CrossRef]
- Zhu, Z.; Labbe, S.; Pena, M.M.; Thiele, D.J. Copper differentially regulates the activity and degradation of yeast Mac1 transcription factor. J. Biol. Chem. 1998, 273, 1277–1280. [Google Scholar] [CrossRef]
- Eliahu, N.; Igbaria, A.; Rose, M.S.; Horwitz, B.A.; Lev, S. Melanin biosynthesis in the maize pathogen Cochliobolus heterostrophus depends on two mitogen-activated protein kinases, Chk1 and Mps1, and the transcription factor Cmr1. Eukaryot. Cell 2007, 6, 421–429. [Google Scholar] [CrossRef]
- Zhao, S.F.; Jiang, H.; Chi, Z.; Liu, G.L.; Chi, Z.M.; Chen, T.J.; Yang, G.; Hu, Z. Genome sequencing of Aureobasidium pullulans P25 and overexpression of a glucose oxidase gene for hyper-production of Ca2+-gluconic acid. Antonie Van Leeuwenhoek 2019, 112, 669–678. [Google Scholar] [CrossRef]
- Xue, S.J.; Chi, Z.; Zhang, Y.; Li, Y.F.; Liu, G.L.; Jiang, H.; Hu, Z.; Chi, Z.M. Fatty acids from oleaginous yeasts and yeast-like fungi and their potential applications. Crit. Rev. Biotechnol. 2018, 38, 1049–1060. [Google Scholar] [CrossRef]
- Qi, C.Y.; Jia, S.L.; Liu, G.L.; Chen, L.; Wei, X.; Hu, Z.; Chi, Z.M.; Chi, Z. Polymalate (PMA) biosynthesis and its molecular regulation in Aureobasidium spp. Int. J. Biol. Macromol. 2021, 174, 512–518. [Google Scholar] [CrossRef]
- Ma, Z.C.; Fu, W.J.; Liu, G.L.; Wang, Z.P.; Chi, Z.M. High-level pullulan production by Aureobasidium pullulans var. melanogenium P16 isolated from mangrove system. Appl. Microbiol. Biotechnol. 2014, 98, 4865–4873. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).