

Whole Genome Sequencing Analysis of Effects of CRISPR/Cas9 in Komagataella phaffii: A Budding Yeast in Distress

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of Target Genes/Regions & Guide RNA Design
2.2. Strains and Constructs
2.2.1. Platform Strains
2.2.2. CRISPR/Cas9 Plasmids
2.3. Transformation and Screening
2.4. Genome Sequencing and Analysis
2.5. Detection of CRISPR/Cas9 Off-Targets
2.6. Transcriptome Sequencing and Analysis
2.7. Growth Analysis
3. Results
3.1. On-Target Behavior
3.1.1. Single Target Transformations
3.1.2. Double Target Transformations
3.1.3. Double Target Transformations within a Non-Essential Region
3.2. Off-Targeting Effects
4. Discussion
4.1. On-Target Behavior
4.2. Off-Targeting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Werten, M.W.T.; Eggink, G.; Cohen Stuart, M.A.; de Wolf, F.A. Production of Protein-Based Polymers in Pichia pastoris. Biotechnol. Adv. 2019, 37, 642–666. [Google Scholar] [CrossRef] [PubMed]
- de Jong, B.; Siewers, V.; Nielsen, J. Systems Biology of Yeast: Enabling Technology for Development of Cell Factories for Production of Advanced Biofuels. Curr. Opin. Biotechnol. 2012, 23, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.V.; Yilma, M.A.; Feliz, A.; Majid, M.T.; Maffetone, N.; Walker, J.R.; Kim, E.; Cho, H.J.; Reynolds, J.M.; Song, M.C.; et al. A Review of the Microbial Production of Bioactive Natural Products and Biologics. Front. Microbiol. 2019, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Davy, A.M.; Kildegaard, H.F.; Andersen, M.R. Cell Factory Engineering. Cell Syst. 2017, 4, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Zhang, Y.; Shi, S. Development and Application of CRISPR/Cas in Microbial Biotechnology. Front. Bioeng. Biotechnol. 2020, 8, 711. [Google Scholar] [CrossRef]
- Weninger, A.; Fischer, J.E.; Raschmanová, H.; Kniely, C.; Vogl, T.; Glieder, A. Expanding the CRISPR/Cas9 Toolkit for Pichia pastoris with Efficient Donor Integration and Alternative Resistance Markers. J. Cell. Biochem. 2018, 119, 3183–3198. [Google Scholar] [CrossRef]
- Cai, P.; Gao, J.; Zhou, Y. CRISPR-Mediated Genome Editing in Non-Conventional Yeasts for Biotechnological Applications. Microb. Cell Fact. 2019, 18, 63. [Google Scholar] [CrossRef]
- David, F.; Siewers, V.; Alper, E.H. Advances in Yeast Genome Engineering. FEMS Yeast Res. 2015, 15, 1–14. [Google Scholar] [CrossRef]
- Gao, S.; Tong, Y.; Wen, Z.; Zhu, L.; Ge, M.; Chen, D.; Jiang, Y.; Yang, S. Multiplex Gene Editing of the Yarrowia lipolytica Genome Using the CRISPR-Cas9 System. J. Ind. Microbiol. Biotechnol. 2016, 43, 1085–1093. [Google Scholar] [CrossRef]
- Rainha, J.; Rodrigues, J.L.; Rodrigues, L.R. CRISPR-Cas9: A Powerful Tool to Efficiently Engineer Saccharomyces cerevisiae. Life 2020, 11, 13. [Google Scholar] [CrossRef]
- Mojica, F.J.M.; Montoliu, L. On the Origin of CRISPR-Cas Technology: From Prokaryotes to Mammals. Trends Microbiol. 2016, 24, 811–820. [Google Scholar] [CrossRef] [PubMed]
- van Overbeek, M.; Capurso, D.; Carter, M.M.; Thompson, M.S.; Frias, E.; Russ, C.; Reece-Hoyes, J.S.; Nye, C.; Gradia, S.; Vidal, B.; et al. DNA Repair Profiling Reveals Nonrandom Outcomes at Cas9-Mediated Breaks. Mol. Cell 2016, 63, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.W.; Arbab, M.; Hsu, J.Y.; Worstell, D.; Culbertson, S.J.; Krabbe, O.; Cassa, C.A.; Liu, D.R.; Gifford, D.K.; Sherwood, R.I. Predictable and Precise Template-Free CRISPR Editing of Pathogenic Variants. Nature 2018, 563, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Dai, Z.; Wang, Q. Advances and Opportunities of CRISPR/Cas Technology in Bioengineering Non-Conventional Yeasts. Front. Bioeng. Biotechnol. 2021, 9, 942. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; McKenna, A.; Schreiber, J.; Haeussler, M.; Yin, Y.; Agarwal, V.; Noble, W.S.; Shendure, J. Massively Parallel Profiling and Predictive Modeling of The Outcomes of CRISPR/Cas9-Mediated Double-Strand Break Repair. Nucleic Acids Res. 2019, 47, 7989–8003. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE Nuclease Architecture for Efficient Genome Editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Klug, A. The Discovery of Zinc Fingers and Their Development for Practical Applications in Gene Regulation and Genome Manipulation. Q. Rev. Biophys. 2010, 43, 1–21. [Google Scholar] [CrossRef]
- Hendel, A.; Kildebeck, E.J.; Fine, E.J.; Clark, J.T.; Punjya, N.; Sebastiano, V.; Bao, G.; Porteus, M.H. Quantifying Genome-Editing Outcomes at Endogenous Loci with SMRT Sequencing. Cell Rep. 2014, 7, 293–305. [Google Scholar] [CrossRef]
- Veres, A.; Gosis, B.S.; Ding, Q.; Collins, R.; Ragavendran, A.; Brand, H.; Erdin, S.; Cowan, C.A.; Talkowski, M.E.; Musunuru, K. Low Incidence of Off-Target Mutations in Individual CRISPR-Cas9 and TALEN Targeted Human Stem Cell Clones Detected by Whole-Genome Sequencing. Cell Stem Cell 2014, 15, 27–30. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Wang, S.; Ren, S.; Bai, R.; Xiao, P.; Zhou, Q.; Zhou, Y.; Zhou, Z.; Niu, Y.; Ji, W.; Chen, Y. No Off-Target Mutations in Functional Genome Regions of a CRISPR/Cas9-Generated Monkey Model of Muscular Dystrophy. J. Biol. Chem. 2018, 293, 11654–11658. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-X.; Opulente, D.A.; Kominek, J.; Zhou, X.; Steenwyk, J.L.; Buh, K.V.; Haase, M.A.B.; Wisecaver, J.H.; Wang, M.; Doering, D.T.; et al. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 2018, 175, 1533–1545.e20. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Burgio, G.; Adams, D.J.; Iyer, V. Collateral Damage and CRISPR Genome Editing. PLoS Genet. 2019, 15, e1007994. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of Double-Strand Breaks Induced by CRISPR–Cas9 Leads to Large Deletions and Complex Rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Höijer, I.; Emmanouilidou, A.; Östlund, R.; van Schendel, R.; Bozorgpana, S.; Tijsterman, M.; Feuk, L.; Gyllensten, U.; den Hoed, M.; Ameur, A. CRISPR-Cas9 Induces Large Structural Variants at on-Target and off-Target Sites in Vivo That Segregate across Generations. Nat. Commun. 2022, 13, 627. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-Seq Enables Genome-Wide Profiling of off-Target Cleavage by CRISPR-Cas Nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef]
- Smith, C.; Gore, A.; Yan, W.; Abalde-Atristain, L.; Li, Z.; He, C.; Wang, Y.; Brodsky, R.A.; Zhang, K.; Cheng, L.; et al. Whole-Genome Sequencing Analysis Reveals High Specificity of CRISPR/Cas9 and TALEN-Based Genome Editing in Human IPSCs. Cell Stem Cell 2014, 15, 12–13. [Google Scholar] [CrossRef]
- Luo, X.; He, Y.; Zhang, C.; He, X.; Yan, L.; Li, M.; Hu, T.; Hu, Y.; Jiang, J.; Meng, X.; et al. Trio Deep-Sequencing Does Not Reveal Unexpected off-Target and on-Target Mutations in Cas9-Edited Rhesus Monkeys. Nat. Commun. 2019, 10, 5525. [Google Scholar] [CrossRef]
- Rayner, E.; Durin, M.-A.; Thomas, R.; Moralli, D.; O’Cathail, S.M.; Tomlinson, I.; Green, C.M.; Lewis, A. CRISPR-Cas9 Causes Chromosomal Instability and Rearrangements in Cancer Cell Lines, Setectable by Cytogenetic Methods. Cris. J. 2019, 2, 406–416. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-Target Effects in CRISPR/Cas9-Mediated Genome Engineering. Mol. Ther.-Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Duan, J.; Lu, G.; Xie, Z.; Lou, M.; Luo, J.; Guo, L.; Zhang, Y. Genome-Wide Identification of CRISPR/Cas9 off-Targets in Human Genome. Cell Res. 2014, 24, 1009–1012. [Google Scholar] [CrossRef] [PubMed]
- Iyer, V.; Boroviak, K.; Thomas, M.; Doe, B.; Riva, L.; Ryder, E.; Adams, D.J. No Unexpected CRISPR-Cas9 off-Target Activity Revealed by Trio Sequencing of Gene-Edited Mice. PLOS Genet. 2018, 14, e1007503. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.R.; Haeussler, M.; Watanabe, C.; Janakiraman, V.; Lund, J.; Modrusan, Z.; Stinson, J.; Bei, Q.; Buechler, A.; Yu, C.; et al. CRISPR Off-Target Analysis in Genetically Engineered Rats and Mice. Nat. Methods 2018, 15, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, K.A.; Wu, W.-H.; Colgan, D.F.; Tsang, S.H.; Bassuk, A.G.; Mahajan, V.B. Unexpected Mutations after CRISPR–Cas9 Editing in Vivo. Nat. Methods 2017, 14, 547–548. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Y.; Zhang, T. Computational Approaches for Effective CRISPR Guide RNA Design and Evaluation. Comput. Struct. Biotechnol. J. 2020, 18, 35–44. [Google Scholar] [CrossRef]
- Jahic, M.; Veide, A.; Charoenrat, T.; Teeri, T.; Enfors, S.O. Process Technology for Production and Recovery of Heterologous Proteins with Pichia pastoris. Biotechnol. Prog. 2006, 22, 1465–1473. [Google Scholar] [CrossRef]
- Bill, R.M. Playing Catch-up with Escherichia coli: Using Yeast to Increase Success Rates in Recombinant Protein Production Experiments. Front. Microbiol. 2014, 5, 1–5. [Google Scholar] [CrossRef]
- Cai, P.; Duan, X.; Wu, X.; Gao, L.; Ye, M.; Zhou, Y.J. Recombination Machinery Engineering Facilitates Metabolic Engineering of the Industrial Yeast Pichia pastoris. Nucleic Acids Res. 2021, 49, 7791–7805. [Google Scholar] [CrossRef]
- Näätsaari, L.; Mistlberger, B.; Ruth, C.; Hajek, T.; Hartner, F.S.; Glieder, A. Deletion of the Pichia pastoris KU70 Homologue Facilitates Platform Strain Generation for Gene Expression and Synthetic Biology. PLoS ONE 2012, 7, e39720. [Google Scholar] [CrossRef]
- Li, P.; Anumanthan, A.; Gao, X.-G.; Ilangovan, K.; Suzara, V.V.; Düzgüneş, N.; Renugopalakrishnan, V. Expression of Recombinant Proteins in Pichia pastoris. Appl. Biochem. Biotechnol. 2007, 142, 105–124. [Google Scholar] [CrossRef]
- Pan, R.; Zhang, J.; Shen, W.L.; Tao, Z.Q.; Li, S.P.; Yan, X. Sequential Deletion of Pichia pastoris Genes by a Self-Excisable Cassette. FEMS Yeast Res. 2011, 11, 292–298. [Google Scholar] [CrossRef]
- Vogl, T.; Sturmberger, L.; Kickenweiz, T.; Wasmayer, R.; Schmid, C.; Hatzl, A.M.; Gerstmann, M.A.; Pitzer, J.; Wagner, M.; Thallinger, G.G.; et al. A Toolbox of Diverse Promoters Related to Methanol Utilization: Functionally Verified Parts for Heterologous Pathway Expression in Pichia pastoris. ACS Synth. Biol. 2016, 5, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Kickenweiz, T.; Pitzer, J.; Sturmberger, L.; Weninger, A.; Biggs, B.W.; Köhler, E.-M.; Baumschlager, A.; Fischer, J.E.; Hyden, P.; et al. Engineered Bidirectional Promoters Enable Rapid Multi-Gene Co-Expression Optimization. Nat. Commun. 2018, 9, 3589. [Google Scholar] [CrossRef] [PubMed]
- Garrigós-Martínez, J.; Vuoristo, K.; Nieto-Taype, M.A.; Tähtiharju, J.; Uusitalo, J.; Tukiainen, P.; Schmid, C.; Tolstorukov, I.; Madden, K.; Penttilä, M.; et al. Bioprocess Performance Analysis of Novel Methanol-Independent Promoters for Recombinant Protein Production with Pichia pastoris. Microb. Cell Fact. 2021, 20, 74. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Fischer, J.E.; Hyden, P.; Wasmayer, R.; Sturmberger, L.; Glieder, A. Orthologous Promoters from Related Methylotrophic Yeasts Surpass Expression of Endogenous Promoters of Pichia pastoris. AMB Express 2020, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Zou, C.; Lin, Y.; Zhang, X.; Ye, Y. Identification and Characterization of PGCW14: A Novel, Strong Constitutive Promoter of Pichia pastoris. Biotechnol. Lett. 2013, 35, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Winkler, C.M.; Kolmbauer, M.; Pichler, H.; Schwab, H.; Emmerstorfer-Augustin, A. Pichia pastoris Protease-deficient and Auxotrophic Strains Generated by a Novel, User-friendly Vector Toolbox for Gene Deletion. Yeast 2019, 36, 557–570. [Google Scholar] [CrossRef]
- Weninger, A.; Hatzl, A.-M.; Schmid, C.; Vogl, T.; Glieder, A. Combinatorial Optimization of CRISPR/Cas9 Expression Enables Precision Genome Engineering in the Methylotrophic Yeast Pichia pastoris. J. Biotechnol. 2016, 235, 139–149. [Google Scholar] [CrossRef]
- Dalvie, N.C.; Leal, J.; Whittaker, C.A.; Yang, Y.; Brady, J.R.; Love, K.R.; Christopher Love, J. Host-Informed Expression of CRISPR Guide RNA for Genomic Engineering in Komagataella phaffii. ACS Synth. Biol. 2020, 9, 26–35. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, G.; Chen, X.; Liu, M.; Zhan, C.; Liu, X.; Bai, Z. High Efficiency CRISPR/Cas9 Genome Editing System with an Eliminable Episomal SgRNA Plasmid in Pichia pastoris. Enzyme Microb. Technol. 2020, 138, 109556. [Google Scholar] [CrossRef]
- Naranjo, C.A.; Jivan, A.D.; Vo, M.N.; de Sa Campos, K.H.; Deyarmin, J.S.; Hekman, R.M.; Uribe, C.; Hang, A.; Her, K.; Fong, M.M.; et al. Role of BGS13 in the Secretory Mechanism of Pichia pastoris. Appl. Environ. Microbiol. 2019, 85, 19. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Weaver, J.; de Sa Campos, K.; Bulahan, R.; Nguyen, J.; Grove, H.; Huang, A.; Low, L.; Tran, N.; Gomez, S.; et al. Mutant Strains of Pichia pastoris with Enhanced Secretion of Recombinant Proteins. Biotechnol. Lett. 2013, 35, 1925–1935. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, K.S.; Irie, K.; Gotoh, Y.; Watanabe, Y.; Araki, H.; Nishida, E.; Matsumoto, K.; Levin, D.E. A Yeast Mitogen-Activated Protein Kinase Homolog (Mpk1p) Mediates Signalling by Protein Kinase C. Mol. Cell. Biol. 1993, 13, 3067–3075. [Google Scholar] [CrossRef] [PubMed]
- Sussman, A.; Huss, K.; Chio, L.-C.; Heidler, S.; Shaw, M.; Ma, D.; Zhu, G.; Campbell, R.M.; Park, T.-S.; Kulanthaivel, P.; et al. Discovery of Cercosporamide, a Known Antifungal Natural Product, as a Selective Pkc1 Kinase Inhibitor through High-Throughput Screening. Eukaryot. Cell 2004, 3, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.E.; Thorner, J. Function and Regulation in MAPK Signaling Pathways: Lessons Learned from the Yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2007, 1773, 1311–1340. [Google Scholar] [CrossRef]
- Sun, Y.; Taniguchi, R.; Tanoue, D.; Yamaji, T.; Takematsu, H.; Mori, K.; Fujita, T.; Kawasaki, T.; Kozutsumi, Y. Sli2 (Ypk1), a Homologue of Mammalian Protein Kinase SGK, Is a Downstream Kinase in the Sphingolipid-Mediated Signaling Pathway of Yeast. Mol. Cell. Biol. 2000, 20, 4411–4419. [Google Scholar] [CrossRef]
- Klis, F.M.; Mol, P.; Hellingwerf, K.; Brul, S. Dynamics of Cell Wall Structure in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2002, 26, 239–256. [Google Scholar] [CrossRef]
- Grossmann, G.; Malinsky, J.; Stahlschmidt, W.; Loibl, M.; Weig-Meckl, I.; Frommer, W.B.; Opekarová, M.; Tanner, W. Plasma Membrane Microdomains Regulate Turnover of Transport Proteins in Yeast. J. Cell Biol. 2008, 183, 1075–1088. [Google Scholar] [CrossRef]
- Cleves, A.E.; Cooper, D.N.; Barondes, S.H.; Kelly, R.B. A New Pathway for Protein Export in Saccharomyces cerevisiae. J. Cell Biol. 1996, 133, 1017–1026. [Google Scholar] [CrossRef]
- Valli, M.; Tatto, N.E.; Peymann, A.; Gruber, C.; Landes, N.; Ekker, H.; Thallinger, G.G.; Mattanovich, D.; Gasser, B.; Graf, A.B. Curation of the Genome Annotation of Pichia pastoris (Komagataella phaffii) CBS7435 from Gene Level to Protein Function. FEMS Yeast Res. 2016, 16, fow051. [Google Scholar] [CrossRef]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces Genome Database: The Genomics Resource of Budding Yeast. Nucleic Acids Res. 2012, 40, D700–D705. [Google Scholar] [CrossRef] [PubMed]
- Sturmberger, L.; Chappell, T.; Geier, M.; Krainer, F.; Day, K.J.; Vide, U.; Trstenjak, S.; Schiefer, A.; Richardson, T.; Soriaga, L.; et al. Refined Pichia pastoris Reference Genome Sequence. J. Biotechnol. 2016, 235, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic Assembly of DNA Molecules up to Several Hundred Kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Kickenweiz, T.; Strumberger, L.; Glieder, A. Bidirectional Promoter. United States Patent Application 2015/0011407 A1, 19 August 2015. [Google Scholar]
- Steiner, S.; Philippsen, P. Sequence and Promoter Analysis of the Highly Expressed TEF Gene of the Filamentous Fungus Ashbya Gossypii. Mol. Gen. Genet. 1994, 242, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Salamanca, J.L.R.D.; Salamanco, M.A.S.G.; Waldsee, M.P.; Neuhofen, H.S. Promoter from Ashbya gossypii. U.S. Patent 6376216 B1, 3 October 2002. [Google Scholar]
- Lin-Cereghino, J.; Wong, W.W.; Xiong, S.; Giang, W.; Luong, L.T.; Vu, J.; Johnson, S.D.; Lin-Cereghino, G.P. Condensed Protocol for Competent Cell Preparation and Transformation of the Methylotrophic Yeast Pichia pastoris. Biotechniques 2005, 38, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Lin-Cereghino, G.P.; Stark, C.M.; Kim, D.; Chang, J.; Shaheen, N.; Poerwanto, H.; Agari, K.; Moua, P.; Low, L.K.; Tran, N.; et al. The Effect of α-Mating Factor Secretion Signal Mutations on Recombinant Protein Expression in Pichia pastoris. Gene 2013, 519, 311. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Suen, W.C.; Windsor, W.; Xiao, L.; Madison, V.; Zaks, A. Improving Tolerance of Candida antarctica Lipase B towards Irreversible Thermal Inactivation through Directed Evolution. Protein Eng. 2003, 16, 599–605. [Google Scholar] [CrossRef]
- Krainer, F.W.; Dietzsch, C.; Hajek, T.; Herwig, C.; Spadiut, O.; Glieder, A. Recombinant Protein Expression in Pichia pastoris Strains with an Engineered Methanol Utilization Pathway. Microb. Cell Fact. 2012, 11, 22. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Küberl, A.; Schneider, J.; Thallinger, G.G.; Anderl, I.; Wibberg, D.; Hajek, T.; Jaenicke, S.; Brinkrolf, K.; Goesmann, A.; Szczepanowski, R.; et al. High-Quality Genome Sequence of Pichia pastoris CBS7435. J. Biotechnol. 2011, 154, 312–320. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinforma. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Cameron, D.L.; Schröder, J.; Penington, J.S.; Do, H.; Molania, R.; Dobrovic, A.; Speed, T.P.; Papenfuss, A.T. GRIDSS: Sensitive and Specific Genomic Rearrangement Detection Using Positional de Bruijn Graph Assembly. Genome Res. 2017, 27, 2050–2060. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly via Adaptive k-Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Khelik, K.; Lagesen, K.; Sandve, G.K.; Rognes, T.; Nederbragt, A.J. NucDiff: In-Depth Characterization and Annotation of Differences between Two Sets of DNA Sequences. BMC Bioinform. 2017, 18, 338. [Google Scholar] [CrossRef]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR Web Toolbox beyond Genome Editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 21 October 2020).
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open Software Development for Computational Biology and Bioinformatics. Genome Biol. 2004, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Pagès, H.; Aboyoun, P.; Gentleman, R.C.; DebRoy, S. Biostrings: Efficient Manipulation of Biological Strings. Available online: https://bioconductor.org/packages/Biostrings (accessed on 7 September 2017).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Sprouffske, K.; Wagner, A. Growthcurver: An R Package for Obtaining Interpretable Metrics from Microbial Growth Curves. BMC Bioinformatics 2016, 17, 172. [Google Scholar] [CrossRef] [PubMed]
- Marobbio, C.M.T.; Agrimi, G.; Lasorsa, F.M.; Palmieri, F. Identification and Functional Reconstitution of Yeast Mitochondrial Carrier for S-Adenosylmethionine. EMBO J. 2003, 22, 5975–5982. [Google Scholar] [CrossRef]
- Zhu, J.; Gong, R.; Zhu, Q.; He, Q.; Xu, N.; Xu, Y.; Cai, M.; Zhou, X.; Zhang, Y.; Zhou, M. Genome-Wide Determination of Gene Essentiality by Transposon Insertion Sequencing in Yeast Pichia pastoris. Sci. Rep. 2018, 8, 10223. [Google Scholar] [CrossRef] [PubMed]
- De Schutter, K.; Lin, Y.-C.; Tiels, P.; Van Hecke, A.; Glinka, S.; Weber-Lehmann, J.; Rouzé, P.; Van de Peer, Y.; Callewaert, N. Genome Sequence of the Recombinant Protein Production Host Pichia pastoris. Nat. Biotechnol. 2009, 27, 561–566. [Google Scholar] [CrossRef]
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Tseng, S.F.; Gabriel, A.; Teng, S.C. Proofreading Activity of DNA Polymerase Pol2 Mediates 3′-End Processing during Nonhomologous End Joining in Yeast. PLoS Genet. 2008, 4, e1000060. [Google Scholar] [CrossRef]
- Wilson, T.E.; Lieber, M.R. Efficient Processing of DNA Ends during Yeast Nonhomologous End Joining: EVIDENCE FOR A DNA POLYMERASE β (POL4 )-DEPENDENT PATHWAY. J. Biol. Chem. 1999, 274, 23599–23609. [Google Scholar] [CrossRef]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef]
- Villarreal, D.D.; Lee, K.; Deem, A.; Shim, E.Y.; Malkova, A.; Lee, S.E. Microhomology Directs Diverse DNA Break Repair Pathways and Chromosomal Translocations. PLoS Genet. 2012, 8, e1003026. [Google Scholar] [CrossRef] [PubMed]
- Seol, J.H.; Shim, E.Y.; Lee, S.E. Microhomology-Mediated End Joining: Good, Bad and Ugly. Mutat. Res. Mol. Mech. Mutagen. 2018, 809, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Schwarzhans, J.P.; Wibberg, D.; Winkler, A.; Luttermann, T.; Kalinowski, J.; Friehs, K. Non-Canonical Integration Events in Pichia pastoris Encountered during Standard Transformation Analysed with Genome Sequencing. Sci. Rep. 2016, 6, 38952. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Ji, J.-H.; Yoon, K.; Che, J.; Seol, J.-H.; Lee, S.E.; Shim, E.Y. Microhomology Selection for Microhomology Mediated End Joining in Saccharomyces cerevisiae. Genes 2019, 10, 284. [Google Scholar] [CrossRef]
- Bae, S.; Kweon, J.; Kim, H.S.; Kim, J.-S. Microhomology-Based Choice of Cas9 Nuclease Target Sites. Nat. Methods 2014, 11, 705–706. [Google Scholar] [CrossRef]
- Decottignies, A. Microhomology-Mediated End Joining in Fission Yeast Is Repressed by Pku70 and Relies on Genes Involved in Homologous Recombination. Genetics 2007, 176, 1403–1415. [Google Scholar] [CrossRef]
- Przewrocka, J.; Rowan, A.; Rosenthal, R.; Kanu, N.; Swanton, C. Unintended On-Target Chromosomal Instability Following CRISPR/Cas9 Single Gene Targeting. Ann. Oncol. 2020, 31, 1270–1273. [Google Scholar] [CrossRef]
- Yan, X.; Li, C.; Yang, J.; Wang, L.; Jiang, C.; Wei, W. Induction of Telomere-Mediated Chromosomal Truncation and Behavior of Truncated Chromosomes in Brassica napus. Plant J. 2017, 91, 700–713. [Google Scholar] [CrossRef]
- Yu, W.; Lamb, J.C.; Han, F.; Birchler, J.A. Telomere-Mediated Chromosomal Truncation in Maize. Proc. Natl. Acad. Sci. USA 2006, 103, 17331–17336. [Google Scholar] [CrossRef]
- Marx, H.; Mecklenbräuker, A.; Gasser, B.; Sauer, M.; Mattanovich, D. Directed Gene Copy Number Amplification in Pichia pastoris by Vector Integration into the Ribosomal DNA Locus. FEMS Yeast Res. 2009, 9, 1260–1270. [Google Scholar] [CrossRef]
- Obenchain, V.; Lawrence, M.; Carey, V.; Gogarten, S.; Shannon, P.; Morgan, M. VariantAnnotation: A Bioconductor package for exploration and annotation of genetic variants. Bioinformatics 2014, 30, 2076–2078. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.L.; Dong, R.; Papenfuss, A.T. StructuralVariantAnnotation: A R/Bioconductor Foundation for A Caller-Agnostic Structural Variant Software Ecosystem. Bioinformatics 2022, 38, 2046–2048. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Madden, T.; Coulouris, G.; Avagyan, V.; Ma, N.; Agarwala, R. BLAST Command Line Applications User Manual. 2008. Available online: https://www.ncbi.nlm.nih.gov/books/NBK279690/ (accessed on 22 April 2022).
- Smith, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013. Available online: http://www.repeatmasker.org (accessed on 22 April 2022).
- Wright, E.S. Using DECIPHER v2.0 to Analyze Big Biological Sequence Data in R. R J. 2016, 8, 352–359. [Google Scholar] [CrossRef]
- Schaper, E.; Kajava, A.V.; Hauser, A.; Anisimova, M. Repeat or not repeat?—Statistical validation of tandem repeat prediction in genomic sequences. Nucleic Acids Res. 2012, 40, 10005. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute. Germline Short Variant Discovery (SNPs + Indels). 2018. Available online: https://software.broadinstitute.org/gatk/best-practices/workflow?id=11145 (accessed on 5 November 2021).







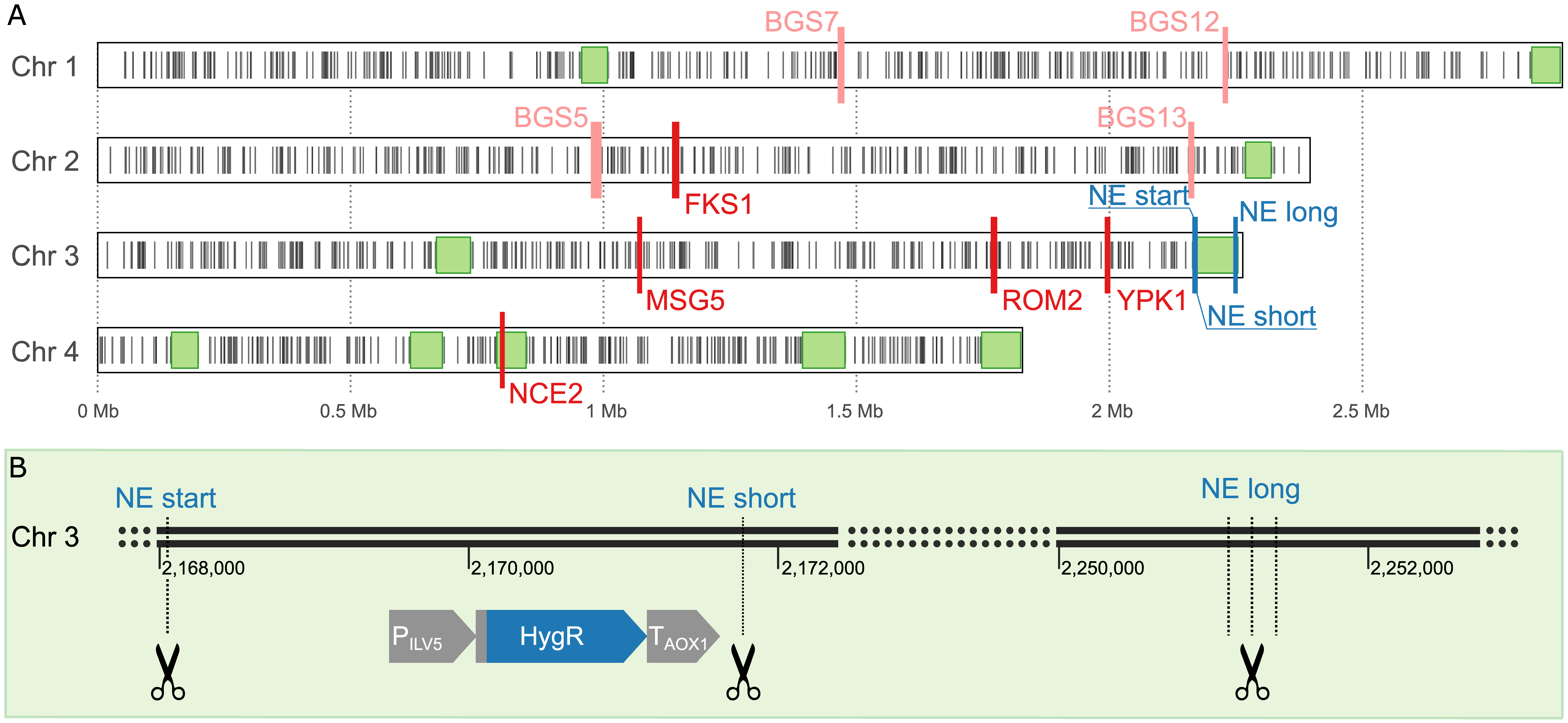{kind=link}
{kind=link}
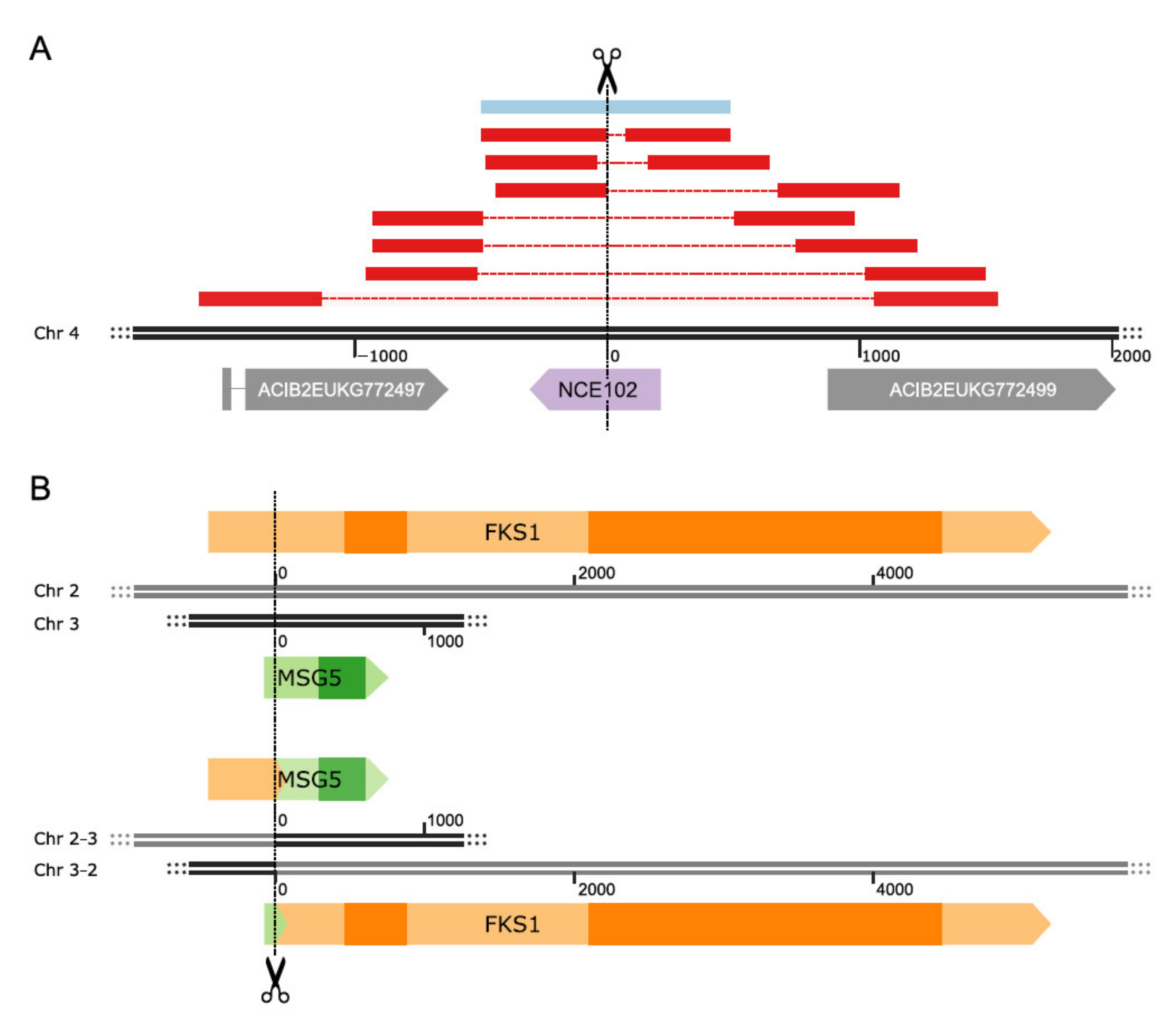{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus Tag | Name | Potential Function/Role | References |
|---|---|---|---|
| ACIB2EUKG769938 | BGS5 | Heavy chain dynein | [52] |
| ACIB2EUKG768596 | BGS7 | Pleckstrin-like, nuclear transport | [52] |
| ACIB2EUKG769034 | BGS12 | Cytoplasmic dynein, intermediate chain | [52] |
| ACIB2EUKG770622 | BGS13 (PKC1) | Pc1 kinase, a protein serine/threonine kinase, which controls a highly conserved signaling pathway managing cell wall integrity | [51,52,53,54] |
| ACIB2EUKG771351 | MSG5 | Dual-specificity protein phosphatase (i.e., Ser/Thr- and Tyr-specific), which plays a role in the regulation of at least two mitogen-activated protein kinase (MAPK)-mediated pathways | [55] |
| ACIB2EUKG771893 | YPK1 | Ser/Thr-protein kinase and is a relevant part of sphingolipid-mediated and cell integrity signaling pathways | [55,56] |
| ACIB2EUKG770030 | FKS1 | 1,3-beta-D-glucan synthase, therefore being responsible for the synthesis of the polysaccharide, which is the main structural component of the cell wall | [57] |
| ACIB2EUKG771759 | ROM2 | One of three guanine nucleotide exchange factors probably specific to Rho1p and Rho2p | [55] |
| ACIB2EUKG772498 | NCE102 | Integral membrane protein, being involved in an alternative pathway for protein export | [58,59] |
| Colony Type | CRIPR Plasmid | Base Strain (#Colonies) | Type | Total Calls | DNMs | DNMs On-Target | DNMs Off-Target | SnpEff Off-Target | CRISPR Off-Target * |
|---|---|---|---|---|---|---|---|---|---|
| single | no gRNA | 3S1K_CalB (1) | SNPs | 16 | 0 | - | 0 | 0 | - |
| InDels | 65 | 0 | - | 0 | 0 | - | |||
| SVs | 93 | 0 | - | 0 | - | - | |||
| single gRNA | UPP-C (6) | SNPs | 91 | 1 | 0 | 1 | 1 | 0 | |
| InDels | 367 | 9 | 6 | 3 | 0 | 1 | |||
| SVs | 224 | 0 | 0 | 0 | - | 0 | |||
| 3S1K_CalB (3) | SNPs | 52 | 2 | 0 | 2 | 2 | 0 | ||
| InDels | 188 | 3 | 2 | 1 | 1 | 0 | |||
| SVs | 190 | 1 | 1 | 0 | - | 0 | |||
| two gRNAs | 3S1K_CalB (9) | SNPs | 161 | 0 | 0 | 0 | 0 | 0 | |
| InDels | 661 | 4 | 4 | 0 | 0 | 0 | |||
| SVs | 733 | 9 | 8 | 1 | - | 0 | |||
| Chr3ne_HygR (2) | SNPs | 33 | 0 | 0 | 0 | 0 | 0 | ||
| InDels | 131 | 1 | 1 | 0 | 0 | 0 | |||
| SVs | 354 | 3 | 1 | 2 | - | 0 | |||
| mixed | no gRNA | 3S1K_CalB (10) | SNPs | 268 | 1 | - | 1 | 0 | - |
| InDels | 209 | 1 | - | 1 | 1 | - | |||
| SVs | 284 | 0 | - | 0 | - | - | |||
| single gRNA | 3S1K_CalB (50) | SNPs | 1935 | 10 | 0 | 10 | 4 | 1 | |
| InDels | 1133 | 31 | 23 | 8 | 6 | 2 | |||
| SVs | 1785 | 18 | 10 | 8 | 1 | ||||
| two gRNAs | 3S1K_CalB (30) | SNPs | 898 | 10 | 0 | 10 | 4 | 3 | |
| InDels | 721 | 34 | 12 | 22 | 19 | 1 | |||
| SVs | 933 | 21 | 15 | 6 | - | 2 | |||
| Chr3ne_HygR (35) | SNPs | 2776 | 24 | 1 | 23 | 16 | 2 | ||
| InDels | 698 | 28 | 16 | 12 | 5 | 3 | |||
| SVs | 1870 | 24 | 18 | 6 | - | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schusterbauer, V.; Fischer, J.E.; Gangl, S.; Schenzle, L.; Rinnofner, C.; Geier, M.; Sailer, C.; Glieder, A.; Thallinger, G.G. Whole Genome Sequencing Analysis of Effects of CRISPR/Cas9 in Komagataella phaffii: A Budding Yeast in Distress. J. Fungi 2022, 8, 992. https://doi.org/10.3390/jof8100992
Schusterbauer V, Fischer JE, Gangl S, Schenzle L, Rinnofner C, Geier M, Sailer C, Glieder A, Thallinger GG. Whole Genome Sequencing Analysis of Effects of CRISPR/Cas9 in Komagataella phaffii: A Budding Yeast in Distress. Journal of Fungi. 2022; 8(10):992. https://doi.org/10.3390/jof8100992
Chicago/Turabian StyleSchusterbauer, Veronika, Jasmin E. Fischer, Sarah Gangl, Lisa Schenzle, Claudia Rinnofner, Martina Geier, Christian Sailer, Anton Glieder, and Gerhard G. Thallinger. 2022. "Whole Genome Sequencing Analysis of Effects of CRISPR/Cas9 in Komagataella phaffii: A Budding Yeast in Distress" Journal of Fungi 8, no. 10: 992. https://doi.org/10.3390/jof8100992
APA StyleSchusterbauer, V., Fischer, J. E., Gangl, S., Schenzle, L., Rinnofner, C., Geier, M., Sailer, C., Glieder, A., & Thallinger, G. G. (2022). Whole Genome Sequencing Analysis of Effects of CRISPR/Cas9 in Komagataella phaffii: A Budding Yeast in Distress. Journal of Fungi, 8(10), 992. https://doi.org/10.3390/jof8100992