Abstract
Setosphaeria turcica is the causal agent of northern corn leaf blight (NCLB), which is a destructive foliar disease of corn around the world. To date, limited information is available on the genetic diversity, population structure, and mating type distribution of the pathogen in the mid-west of China. In this study, based on single nucleotide polymorphism (SNP) markers and mating type-specific primers, we characterized 117 S. turcica isolates collected from Henan, Hebei, Shanxi, and Shaanxi provinces in China. Based on the developed 33 SNP markers, all isolates can be categorized into two genetic groups. Each group consisted of isolates from all four provinces. The Nei’s gene diversity of four populations ranged from 0.328 to 0.419 with a mean of 0.391. The analysis of fixation index (Fst) and gene flow (Nm) suggested that low genetic differentiation and high gene flow existed among four geographic populations. The analysis of molecular variance (AMOVA) demonstrated that the principal molecular variance existed within populations (98%) rather than among populations (2%). The analysis of mating type loci revealed that two mating types (MAT1-1 and MAT1-2) were basically in equilibrium in all four populations. These findings advance our understanding of the genetic diversity, population structure and mating type distribution of S. turcica on corn in the mid-west of China and will aid in developing efficient strategies to control NCLB.
1. Introduction
Corn (Zea mays L.) is one of the most important crops in the world. More than half of its global production is used for livestock feeds and various industrial raw materials [1]. Northern corn leaf blight (NCLB) caused by Setosphaeria turcica (Luttr.) K. J. Leonard & Suggs [anamorph, Exserohilum turcicum (Pass.) K. J. Leonard & Suggs] is a devastating foliar disease worldwide [2], which leads to great losses in corn production. In some circumstances, such as low temperature and humid weather, this pathogen can even cause more than 50% yield reduction [3]. In China, NCLB was first reported in 1899 [4] and the disease typically occurs in most corn-growing areas. S. turcica causes diseases mainly by inducing necrotic lesions that reduce leaf area available for photosynthesis. During infection of corn by S. turcica, leaves first show water-soaked gray-green lesions, and then develop into larger long fusiform necrotic lesions [5]. When the disease is severe, the lesions spots connect with each other, leading to death of the whole plant.
S. turcica is a heterothallic ascomycetous fungus, which possesses two mating types, namely MAT1-1 and MAT1-2. Sexual reproduction occurs when the isolates with opposite mating types are in close proximity [6]. The sexual stage of S. turcica can be induced under laboratory conditions, and it has also been observed on heavily infected corn leaves from natural fields [7]. Though observations of sexual reproduction in nature are rare, it may be one important source driving genetic variation of the fungal pathogen.
Understanding the genetic diversity and population structure of S. turcica is crucial for studying the epidemiology and disease management of NCLB. Various molecular markers, such as random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), sequence-related amplified polymorphism (SRAP), inter-simple sequence repeat (ISSR) and simple sequence repeat (SSR) have been utilized to evaluate the genetic diversity of the pathogen [8,9,10,11,12]. Single nucleotide polymorphism (SNP) is a third-generation molecular marker and has been widely used for studying genetic diversity and population structures of plants and animals, owing to the advantages of ubiquitous presence, uniform distribution, high heritability, and bi-allelic nature [13,14]. However, the SNP marker is rarely applied to plant pathogens at present.
NCLB has occurred seriously and become a major maize disease in the mountainous region of China. Genetic diversity of S. turcica on corn was studied in the north and south of China [2,8,12], but so far studies of genetic diversity have been not performed in the middle of China. In this study, we developed SNP markers for S. turcica on corn. Based on the SNP markers, the genetic diversity and population structure of S. turcica from corn were evaluated in the mid-west of China. In addition, we identified the mating types of S. turcica isolates using specific PCR primers and analyzed the ratio of mating types.
2. Materials and Methods
2.1. Collection and Isolation of S. turcica
The S. turcica isolates used in this study were collected from Henan, Hebei, Shanxi and Shaanxi provinces, China, in September 2019 (Supplementary Figure S1). Of 117 isolates, 30 were from Henan, Hebei, and Shanxi, respectively, and 27 were from Shaanxi (Supplementary Table S1). Fungal samples were isolated by the tissue separation method. Briefly, leaves with typical symptoms (Supplementary Figure S2A) were cut into small pieces, treated with 70% ethanol for 1 min, and soaked in 3% sodium hypochlorite solution for another 2 min. After being rinsed three times with sterile water, the leaf fragments were placed on potato dextrose agar (PDA, HuanKai Microbial, Guangzhou, China), amended with 50 μg/mL rifampin (Solarbio, Beijing, China), and incubated at 25 °C for 3 to 5 days. After being examined by light microscopy (Phenix, Shangrao, China), a single hyphal tip of each isolate was picked for another subculture. Purified isolates (Supplementary Figure S2B–E) were stored at 4 °C on PDA slants.
2.2. DNA Extraction
S. turcica isolates were grown in potato dextrose broth at 25 °C for 3 days. Mycelia of all isolates were collected and dried by sterilized filter papers. DNA extraction was conducted by an Ezup Column Fungi Genomic DNA Purification Kit (Sangon, Shanghai, China) according to the manufacturer’s instructions. The quality and concentration of the gDNA were determined using a NanoDrop 2000 (Thermo Fisher Scientific, Wilmington, DE, USA).
2.3. SNP Marker Development
A total of 83,717 SNP loci were identified from whole genome resequencing data of 15 S. turcica isolates from corn (https://www.ncbi.nlm.nih.gov/sra/PRJNA834655) (accessed on 3 May 2022) using GATK v4.2.0.0 (Aaron McKenna, Cambridge, MA, USA) [15]. We selected 80 candidate SNP loci that showed a minimal distance of 250 kb from each other. The primers were designed by Batchprimer 3 (https://wheat.pw.usda.gov/demos/BatchPrimer3) (accessed on 29 May 2008) with amplicon sizes ranging from 140 to 270 bp. Based on the 80 SNP loci, 76 SNP primer pairs were successfully designed (Supplementary Table S2) and were commercially synthesized by Sangon Biotech Co., Ltd. (Shanghai, China).
2.4. Multiplex PCR and Sequencing
Two panels that contain 76 target SNP sites were designed. One contains 49 target SNP sites, and the other contains 27 target SNP sites. The DNA sequencing library preparation was performed by two-step PCR. Firstly, multiplex PCR was performed based on the two panels. The PCR products were tested by electrophoresis using a 1% (w/v) agarose gel (Sangon, Shanghai, China) in TBE buffer, stained with ethidium bromide (EB) and visualized under UV light to determine the size of PCR products, then PCR products were separated from the gel and recovered using AMPure XP magnetic beads (Beckman Coulter, Brea, CA, USA). The second PCR was performed using the PCR products derived from the first round, universal P5 primer and universal P7 primer to obtain the sequencing library with molecular tags. The amplicons were purified by AMPure XP magnetic beads. The specific steps were described by Cui et al. [16]. Finally, paired-end sequencing of the library was performed on the HiSeq XTen sequencers (Illumina, San Diego, CA, USA).
Raw reads were filtered by removing sequencing adapters and low-quality reads using Cutadapt v1.2.1 (NBIS, Dortmund, Germany) [17] and Prinseq-lite v0.20.3 (SOURCEFORGE, Santiago, MN, USA) [18]. The cleaned reads were mapped to the S. turcica reference genome (GenBank assembly accession: GCA_013390295.1) using BWA v0.7.13-r1126 (Wheeler Aligner, Cambridge, MA, USA) [19] with default parameters. Samtools v0.1.18 (GitHub, Cambridge, MA, USA) [20] was used to calculate the genotype likelihoods of target sites. The alternate allele frequency was greater than 90%, and SNP loci with missing data were excluded.
2.5. Genetic Diversity and Phylogenetic Analysis
In order to analyse the genetic diversity of S. turcica, the number of alleles (Na), effective number of alleles (Ne), allele frequencies, Nei’s gene diversity (h) [21] and Shannon’s polymorphism information index (I) [22] were calculated using the program POPGENE v1.31 (Microsoft, Edmonton, AB, Canada) [23]. Phylogenetic analysis was performed by Mega v7.0 (PubMed, Philadelphia, PA, USA) [24] with 1000 bootstrap replications using the neighbor-joining (NJ) method.
2.6. Genetic Differentiation Analysis
In order to analyse the genetic differentiation between populations, the fixation index (Fst) and gene flow (Nm) were calculated with GenAIEx v6.5.2 (Microsoft, Canberra, Australia) [25]. Analysis of molecular variance (AMOVA) was used to apportion the variation within and between populations using the same software.
2.7. Population Structure Analysis
Structure V2.3.4 (Palo Alto, Santa Clara, CA, USA) [26] was used to analyze the population structure based on an admixture model. Structure was implemented for K = 1 to 10 with 10 replications after an initial burn-in of 100,000 generations followed by a run length of 1,000,000 generations. The most optimal value of K was identified according to Evanno’s method [27], using Structure Harvester [28].
2.8. Mating Type Determination
Mating types of S. turcica isolates were examined by multiple PCR assays on the basis of the mating type-specific primers reported by Haasbroek M.P. [29]. The S. turcica MAT1-1 locus and MAT1-2 locus were amplified using the same reverse primer (MAT_CommonR, 5′-AATGCGGACACGGAATAC-3′). Forward primer MAT_1-1F (5′-CTCGTCCTTGGAGAAGAATATC-3′) was employed for MAT1-1, and MAT_1-2F (5′-GCTCCTGGACCAAATAATACA-3′) for MAT1-2. PCR was performed as described previously [16]. The mating type-specific primer pairs generated 608 bp amplicons of MAT1-1 and 393 bp of MAT1-2. Ratios between MAT1-1 and MAT1-2 from each geographical population were subjected to a χ2 test using IBM SPSS Statistics 20 (International Business Machines Corporation, Arkmonk, NY, USA) to evaluate divergence from the expected ratio of 1:1 at the p < 0.05 level.
3. Results
3.1. Genetic Diversity
Based on 80 candidate SNP loci, 76 primer pairs were successfully designed by Batchprimer 3. A total of 33 primer pairs amplified the expected DNA bands from all 117 isolates, and the 33 SNP loci were polymorphic (Supplementary Table S3). The observed number of alleles at each locus were two, so all SNP markers were biallelic markers (Table 1). The number of effective alleles per locus ranged from 1.246 to 1.999, with a mean of 1.733. The minor allele frequency (MAF) ranged from 0.111 to 0.495, with 70% of the markers having MAF > 0.2. The Nei’s gene diversity showed a range of 0.198 to 0.5, with a mean of 0.406. Shannon’s information index varied from 0.349 to 0.693, with a mean of 0.593. Thirty-three SNP markers were successfully developed for genetic diversity analysis of S. turcica.

Table 1.
Characteristics of 33 SNP markers.
At the population level, the Nei’s gene diversities were 0.419 (Henan), 0.412 (Shaanxi), 0.404 (Hebei) and 0.328 (Shanxi), with an average of 0.391 (Table 2). Shannon’s information indices were 0.625, 0.599, 0.587 and 0.504 for Henan, Shaanxi, Hebei, and Shanxi populations, respectively. The genetic diversity of the Shanxi population was lower than the other three populations, and the genetic diversity of the Henan population was the most abundant.
Table 2.
Genetic diversity of Setosphaeria turcica populations from Henan, Hebei, Shanxi and Shaanxi in China.
3.2. Phylogenetic Analysis
Using the neighbor-joining method, 117 S. turcica isolates were categorized into two groups (Figure 1). There were 60 isolates in group I, including 13 isolates from Henan, 13 isolates from Hebei, 21 isolates from Shanxi, and 13 isolates from Shaanxi. There were 57 isolates in group II, including 17 isolates from Henan, 17 isolates from Hebei, 9 isolates from Shanxi, and 14 isolates from Shaanxi. These isolates from four geographic populations were divided into one group, and even into one clade. These results suggested that the migration of S. turcica could happen frequently among the four provinces in China and there was no significant correlation between genetic groups and geographic origin.
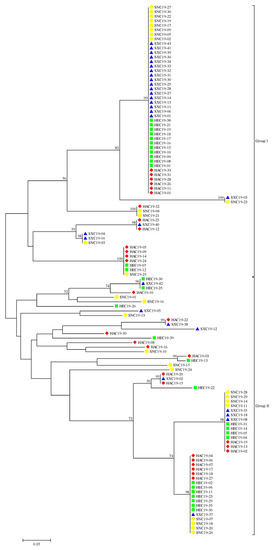
Figure 1.
Phylogenetic tree of 117 Setosphaeria turcica isolates based on the neighbor-joining method. The red rhombus, green square, blue triangle, and yellow circle represent the Henan, Hebei, Shanxi, and Shaanxi populations, respectively.
3.3. Genetic Differentiation
Genetic differentiation among populations was estimated using Fst. The Fst values among the four populations of S. turcica ranged from 0.006 to 0.057 (Table 3), suggesting that there existed some genetic differentiation among the four populations, but the genetic differentiation was relatively low. Gene flow analysis showed that the Nm values among the four populations of S. turcica varied from 6.647 to 45.148 (Table 3). All values of Nm exceeded 6, suggesting that high gene flow existed between the pairwise populations of the four geographic populations. The AMOVA analysis showed that 98% of the total genetic variation was distributed within the four populations (Table 4). The remaining 2% of the genetic variance was caused by geographic separation among the four geographic populations.
Table 3.
Pairwise matrices of Fst (below the diagonal) and Nm (above the diagonal) among four populations of Setosphaeria turcica in China.
Table 4.
Molecular variance analysis within and among four populations of Setosphaeria turcica in China.
3.4. Population Structure
The Delta K value was highest at K = 2, suggesting that two genetic clusters best explained the population structure of S. turcica isolates from four geographic populations in China (Figure 2). Though each population consisted of two genetic clusters in the four geographic populations, the main source of clusters in every population was slightly different. The proportion of the red cluster was more in the Henan population, while the proportion of the green cluster was more in the Shanxi population. The two clusters were relatively evenly distributed in Hebei and Shaanxi populations. These results indicated that there was a low degree of genetic differentiation among the four populations.
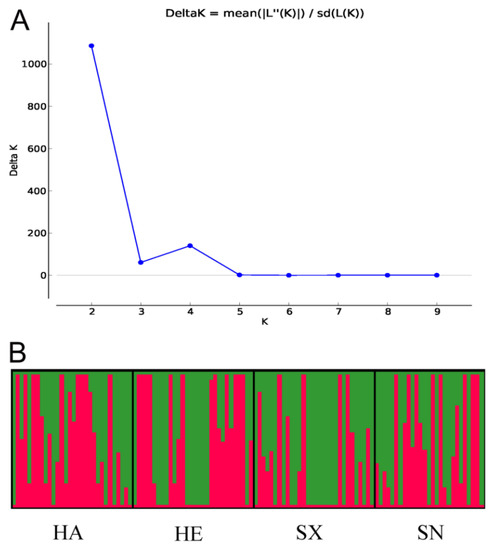
Figure 2.
Graphical representation of the results obtained from STRUCTURE. (A) DeltaK curve showing evidence of two genetic clusters. (B) STRUCTURE plot of 117 Setosphaeria turcica isolates with K = 2. Each vertical line represents an isolate. Boxes framed by thick black lines indicate different geographic populations.
3.5. Mating Type Distribution
Based on the mating type-specific primers, the mating type idiomorph was successfully amplified in all 117 S. turcica isolates and all isolates had only either the MAT1-1 or MAT1-2 mating type idiomorph. Forty-five isolates were determined as MAT1-1, and the remaining 72 isolates were determined as MAT1-2. The ratio of MAT1-1 and MAT1-2 for all isolates did not differ significantly from a 1:1 ratio (p = 0.8, Table 5). For each geographic population, the frequencies of mating type also did not deviate significantly from a 1:1 ratio (0.1 ≤ p ≤ 0.6, Table 5). These results suggested that sexual reproduction of S. turcica could be occurring in the four populations of China.
Table 5.
Mating type distribution in four Chinese populations of Setosphaeria turcica.
4. Discussion
In this study, 33 SNP markers were developed for S. turcica on corn based on 80 SNP loci. In our previous study, 36 SNP markers were developed for S. turcica on sorghum based on 80 SNP loci [16]. Of these SNP loci, 50 SNP loci were identical for the two studies. In the 50 SNP loci, 13 SNP markers were developed for S. turcica on corn and 12 SNP markers were developed for S. turcica on sorghum. Only 7 SNP markers can be used not only for S. turcica on corn but also for S. turcica on sorghum. Six SNP markers can be used only for S. turcica on corn and 5 SNP markers can be used only for S. turcica on sorghum, suggesting that genetic difference between S. turcica on corn and S. turcica on sorghum is significant.
Genetic diversity is the basis of biological diversity research, which reflects the adaptability of populations and the potential to be transformed and utilized [30]. Nei’s gene diversity (h) is an important parameter for measuring genetic diversity of a population [31]. In the present study, Nei’s gene diversity (h) ranged from 0.328 to 0.419 with a mean of 0.391, indicating that the genetic diversity of S. turcica was relatively low in the mid-west of China. In the recent studies, Ma et al. [11] reported that low genetic diversity (h = 0.2634) was detected within the S. turcica populations in the north of China, and Dai et al. [12] reported that the Fujian population of S. turcica showed a low gene diversity (h = 0.13) in the south of China. These findings suggest that there is a low gene diversity in the Chinese populations of S. turcica. The result is consistent with previous findings of genetic diversity of S. turcica in other countries. Human et al. [10] reported low genetic diversity of S. turcica in South Africa, and Turgay et al. [32] also reported low genetic diversity of S. turcica in Turkey.
Gene flow is an important factor that can reduce geographic differentiation and result in the homogenization of genetic characteristics among populations [33]. In our study, a high degree of gene flow was detected among the four geographic populations of S. turcica. Henan, Shaanxi, Hebei, and Shanxi are four neighboring provinces in the mid-west of China. The migration of S. turcica among the four provinces should be main cause of high gene flow, which is supported by the phylogenetic analysis that S. turcica isolates from four geographic populations were divided into one clade. Gene flow homogenizes populations by genetically decreasing variance among populations and increasing variance within populations. So a low level of genetic differentiation was detected among the four geographic populations of S. turcica, which is consistent with the AMOVA analysis that the principal molecular variance existed within populations rather than among populations.
Understanding the distribution of the mating types of a phytopathogen is essential to infer the level of population genetic variability attributable to the underlying sexual recombination [34]. In our study, MAT1-2 was the predominant mating type in the four geographic populations, but the ratio of MAT1-1 and MAT1-2 did not differ significantly from a 1:1 ratio (p < 0.05), implying that sexual reproduction of S. turcica probably occurs under natural conditions in the four provinces of China. Sexual reproduction has been hypothesized as a critical factor causing high genetic variation in plant fungal pathogens [35,36]. However, a relatively low genetic diversity was detected in the four geographic populations. This may be because sexual reproduction of S. turcica occurs at a low frequency and asexual reproduction is still the main reproductive mode. At present, sexual reproduction of S. turcica has not been observed in natural corn fields of China. In future, we will investigate the sexual reproduction of S. turcica in order to figure out its role in Chinese populations.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jof8111165/s1, Figure S1: Map illustrating the location of four provinces in which Setosphaeria turcica isolates were collected. The blue regions denote coastlines or islands. HA = Henan province, HE = Hebei province, SX = Shanxi province, SN = Shaanxi province; Figure S2: Northern corn leaf blight and Setosphaeria turcica. (A) Typical symptoms of northern corn leaf blight. (B) The fungal colony (upper surface). (C) The fungal colony (lower surface). (D) Conidium. (E) Conidiophore; Table S1: Setosphaeria turcica isolates used in this study; Table S2: 76 SNP loci and primer pairs for Setosphaeria turcica. Table S3: 33 SNP loci and primer pairs used to analyze genetic diversity of Setosphaeria turcica populations in China.
Author Contributions
Conceptualization, Z.H. and Y.H.; methodology, L.C., L.Z. and B.W.; software, L.C. and L.Z.; writing—original draft preparation, all authors; writing—review and editing, Z.H. and Y.H.; funding acquisition, L.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Henan Provincial Science and Technology Major Project (project no. 221100110100), the Key Scientific and Technological Project of Henan Province (project no. 212102110141) and the National Science Foundation of China (project no. U1804104).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data may be accessed by contacting the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nieuwoudt, A.; Human, M.P.; Craven, M.; Crampton, B.G. Genetic differentiation in populations of Exserohilum turcicum from maize and sorghum in South Africa. Plant Pathol. 2018, 67, 1483–1491. [Google Scholar] [CrossRef]
- Dong, J.; Fan, Y.; Gui, X.; An, X.; Ma, J.; Dong, Z. Geographic distribution and genetic analysis of physiological races of Setosphaeria turcica in Northern China. Am. J. Agric. Biol. Sci. 2008, 3, 389–398. [Google Scholar]
- Hooda, K.S.; Khokhar, M.K.; Shekhar, M.; Karjagi, C.G.; Kumar, B.; Mallikarjuna, N.; Devlash, R.K.; Chandrashekara, C.; Yadav, O.P. Turcicum leaf blight-sustainable management of a re-emerging maize disease. J. Plant Dis. Protect. 2017, 124, 101–113. [Google Scholar] [CrossRef]
- Chen, G. Distribution and control method of Exserohilum turcicum race 2. J. Maize Sci. 1993, 1, 65–66. [Google Scholar]
- Shi, N.N.; Du, Y.X.; Ruan, H.C.; Yang, X.J.; Dai, Y.L.; Gan, L.; Chen, F.R.; Liu, X.Z. First report of northern corn leaf blight caused by Setosphaeria turcica on corn (Zea mays) in Fujian province, China. Plant Dis. 2017, 101, 831–832. [Google Scholar] [CrossRef]
- Luttrell, E.S. The perfect stage of Helminthosporium turcicum. Phytopathology 1958, 48, 281–287. [Google Scholar]
- Bunkoed, W.; Kasam, S.; Chaijuckam, P.; Yhamsoongnern, J.; Prathuangwong, S. Sexual reproduction of Setosphaeria turcica in natural corn fields in Thailand. Kasetsart J. Nat. Sci. 2014, 48, 175–182. [Google Scholar]
- Fan, Y.; Ma, J.; Gui, X.; An, X.; Sun, S.; Dong, J. Distribution of mating types and genetic diversity induced by sexual recombination in Setosphaeria turcica in Northern China. Front. Agric. China 2007, 1, 368–376. [Google Scholar] [CrossRef]
- Muiru, W.; Koopmann, B.; Tiedemann, A.; Mutitu, E.; Kimenju, J. Evaluation of genetic variability of Kenyan, German and Austrian isolates of Exserohilum turcicum using amplified fragment length polymorphism DNA marker. Biotechnology 2010, 9, 204–211. [Google Scholar] [CrossRef]
- Human, M.P.; Barnes, I.; Craven, M.; Crampton, B.G. Lack of population structure and mixed reproduction modes in Exserohilum turcicum from South Africa. Phytopathology 2016, 106, 1386–1392. [Google Scholar] [CrossRef]
- Ma, Z.; Liu, B.; He, S.; Gao, Z. Analysis of physiological races and genetic diversity of Setosphaeria turcica (Luttr.) K. J. Leonard & Suggs from different regions of China. Can. J. Plant Pathol. 2020, 42, 396–407. [Google Scholar]
- Dai, Y.; Gan, L.; Lan, C.; Lu, X.; Yang, X.; Gao, Z. Genetic differentiation and mixed reproductive strategies in the northern corn leaf blight pathogen Setosphaeria turcica from sweet corn in Fujian province, China. Front. Microbiol. 2021, 12, 632575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Han, W.; Tang, H.; Li, G.; Zhang, M.; Xu, R.; Liu, Y.; Yang, T.; Li, W.; Zou, J.; et al. Genomic diversity dynamics in conserved chicken populations are revealed by genome-wide SNPs. BMC Genom. 2018, 19, 598. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Chhokar, V.; Sheoran, S.; Singh, R.; Tiwari, R. Characterization of genetic diversity and population structure in wheat using array based SNP markers. Mol. Biol. Rep. 2020, 47, 293–306. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Deng, J.; Zhao, L.; Hu, Y.; Liu, T. Genetic diversity and population genetic structure of Setosphaeria turcica from sorghum in three provinces of China using single nucleotide polymorphism markers. Front. Microbiol. 2022, 13, 853202. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. ACM SIGMOBILE Mob. Comput. Commun. Rev. 2001, 5, 3–55. [Google Scholar] [CrossRef]
- Yeh, F.C.; Yang, R.; Boyle, T.B.; Ye, Z.; Mao, J.X. POPGENE, the user-friendly shareware for population genetic analysis. Mol. Biol. Biotechnol. Cent. Univ. Alta. Can. 1997, 10, 295–301. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; VonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Haasbroek, M.P.; Craven, M.; Barnes, I.; Crampton, B.G. Microsatellite and mating type primers for the maize and sorghum pathogen, Exserohilum turcicum. Australas. Plant Pathol. 2014, 43, 577–581. [Google Scholar] [CrossRef]
- Reed, D.H.; Frankham, R. Correlation between fitness and genetic diversity. Conserv. Biol. 2003, 17, 230–237. [Google Scholar] [CrossRef]
- Lu, N.; Zheng, W.; Wang, J.; Zhan, G.; Huang, L.; Kang, Z. SSR analysis of population genetic diversity of Puccinia striiformis f. sp. tritici in Longnan region of Gansu, China. Sci. Agric. Sin. 2009, 42, 2763–2770. [Google Scholar]
- Turgay, E.B.; Çelik Oğuz, A.; Ölmez, F.; Tunalı, B.; Kurt, Ş.; Akçalı, E.; Baran, B.; Büyük, O.; Helvacıoğlu, Ö.; Enginsu, S.; et al. Genetic diversity and mating-type frequency of Exserohilum turcicum in Turkey. J. Phytopathol. 2021, 169, 570–576. [Google Scholar] [CrossRef]
- Slatkin, M. Gene flow and the geographic structure of natural populations. Science 1987, 236, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Valent, B.; Farrall, L.; Chumley, F.G. Magnaporthe grisea genes for pathogenicity and virulence identified through a series of backcrosses. Genetics 1991, 127, 87–101. [Google Scholar] [CrossRef]
- Glass, N.L.; Kuldau, G.A. Mating type and vegetative incompatibility in filamentous ascomycetes. Annu. Rev. Phytopathol. 1992, 30, 201–224. [Google Scholar] [CrossRef]
- Bi, Y.; Hu, J.; Cui, X.; Shao, J.; Lu, X.; Meng, Q.; Liu, X. Sexual reproduction increases the possibility that Phytophthora capsici will develop resistance to dimethomorph in China. Plant Pathol. 2014, 63, 1365–1373. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).