
Study of Dimorphism Transition Mechanism of Tremella fuciformis Based on Comparative Proteomics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungus Strains and Culture Conditions
2.2. Protein Extraction, Digestion, and Peptide Fractions
2.3. HRMS1-DIA-Based LC–MS/MS
2.4. Parallel Reaction Monitoring (PRM) Target Proteomics
2.5. Data Processing and Statistical Analysis
2.6. Bioinformatics Analysis
2.7. Metabolomics Analysis
3. Results
3.1. HRMS1-DIA Quantification of FBMds and DM
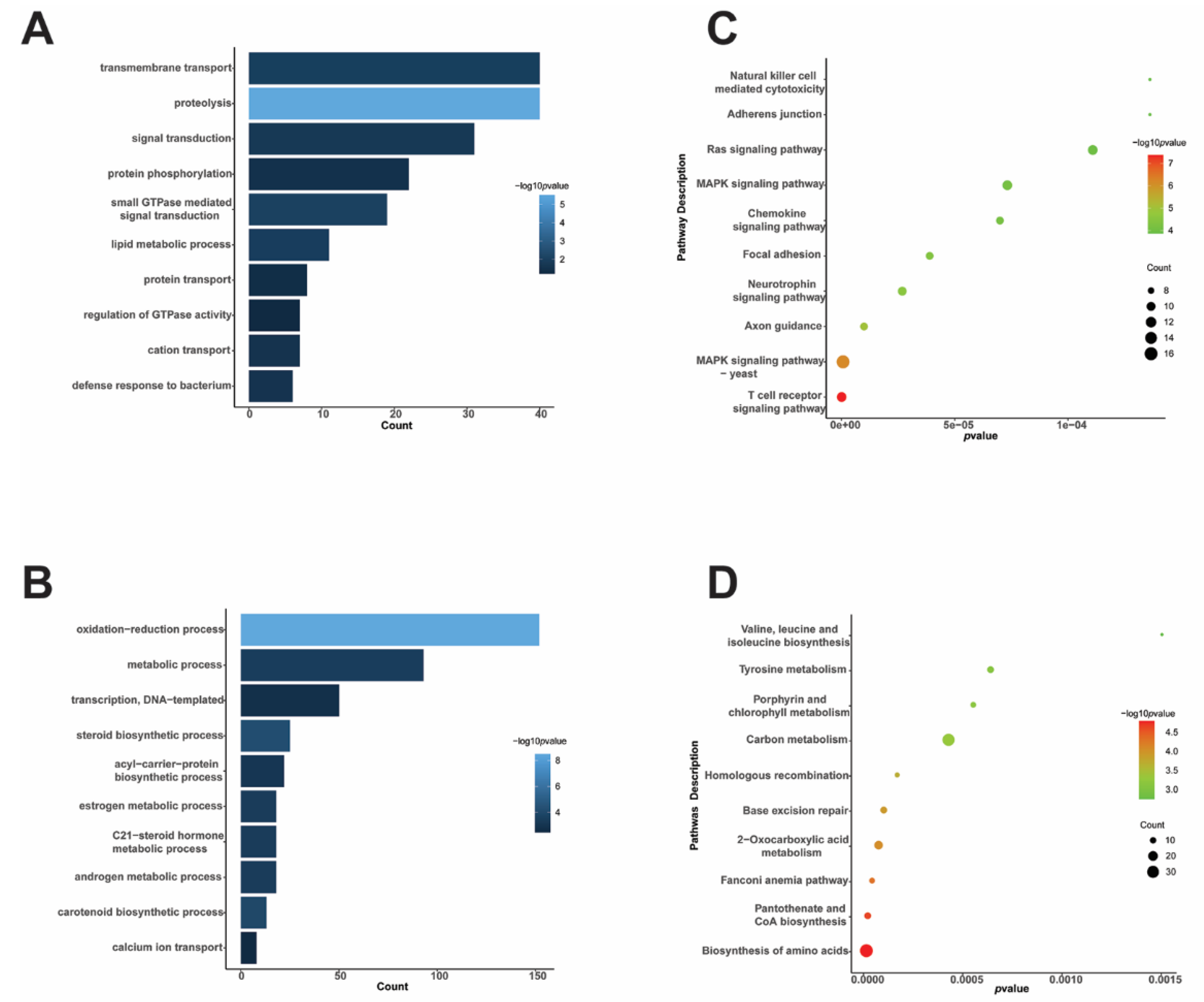
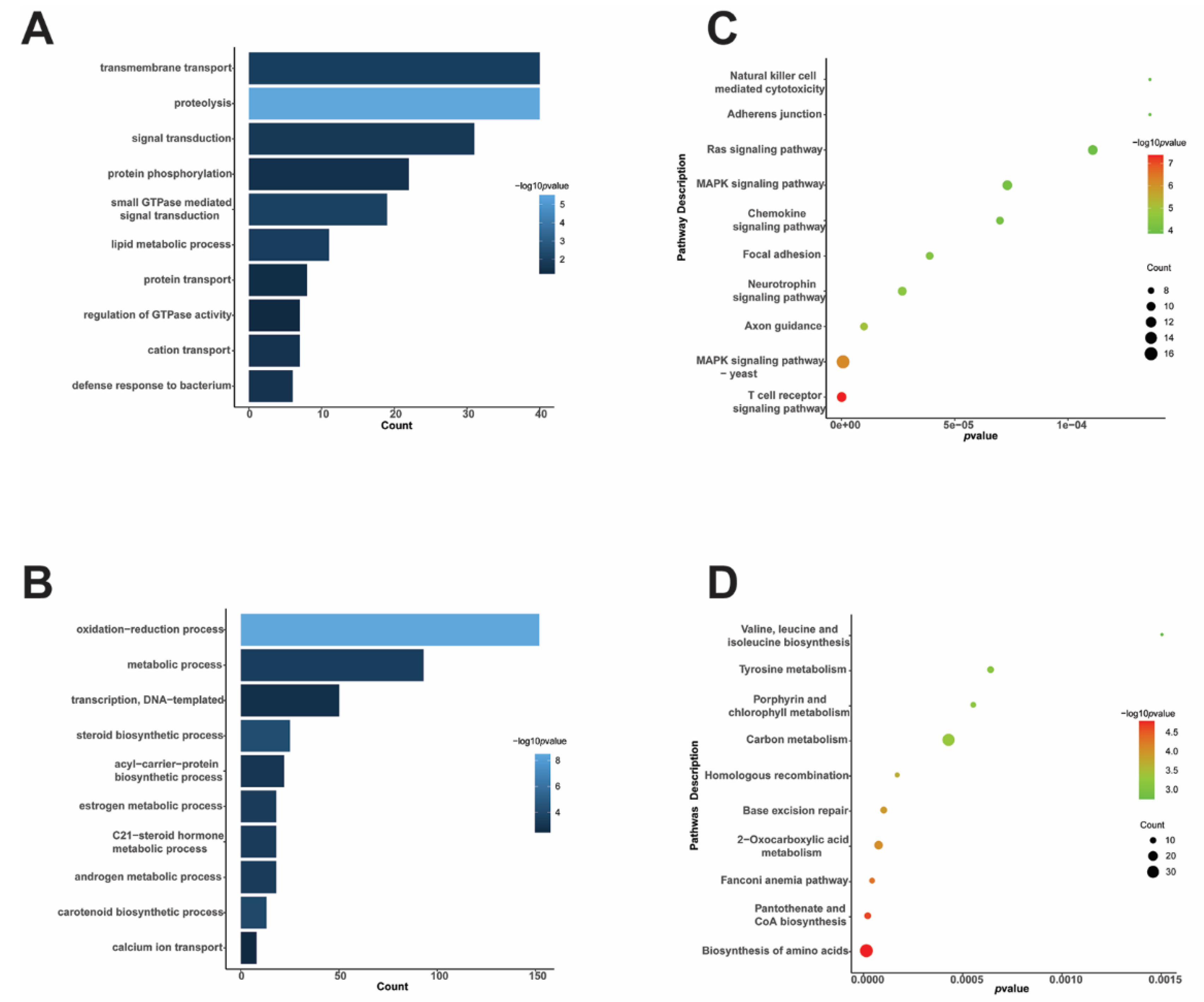
3.2. Functional Analysis of DEPs of T. fuciformis Dimorphism
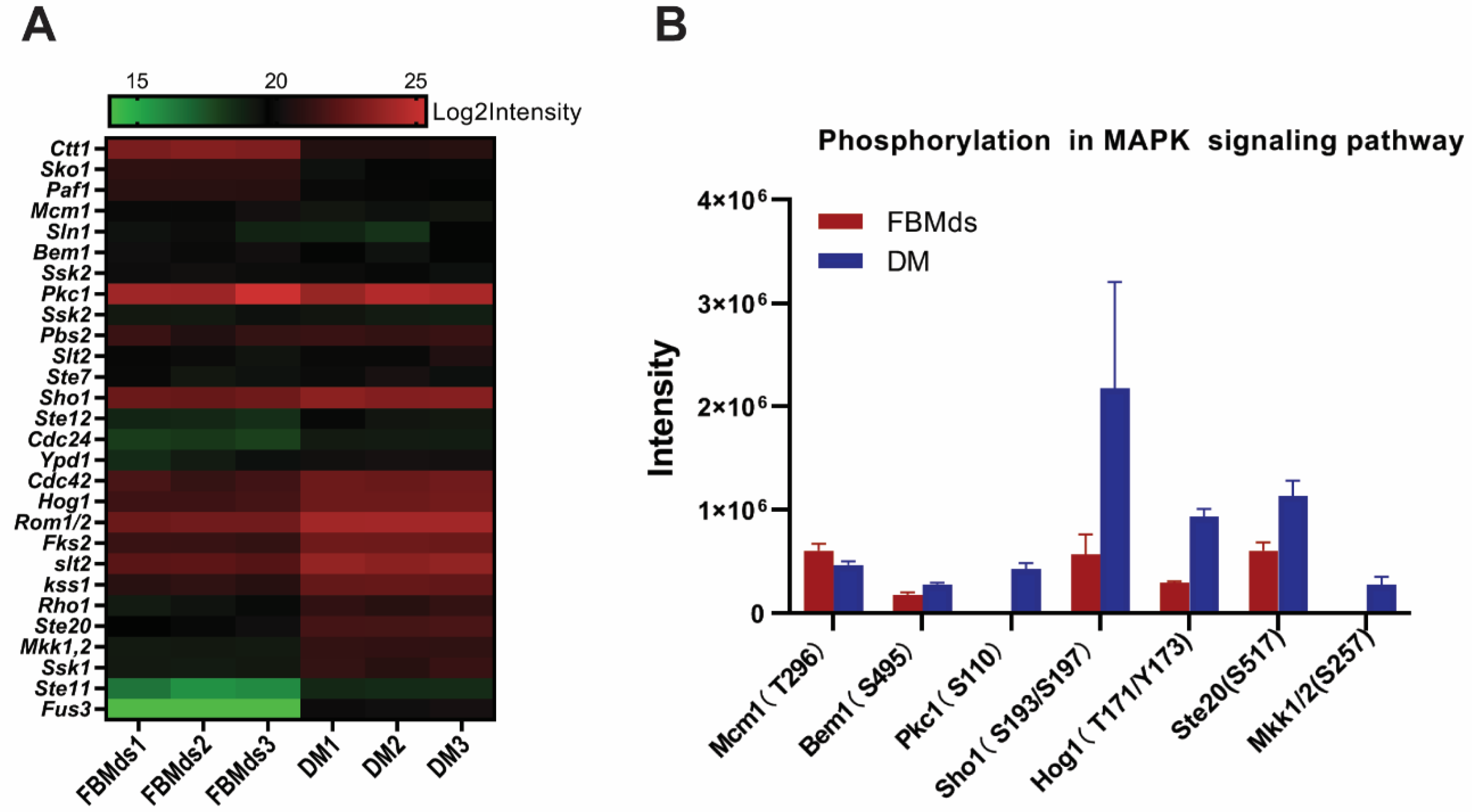
3.3. PRM Validation of HRMS1-DIA Proteomics Results
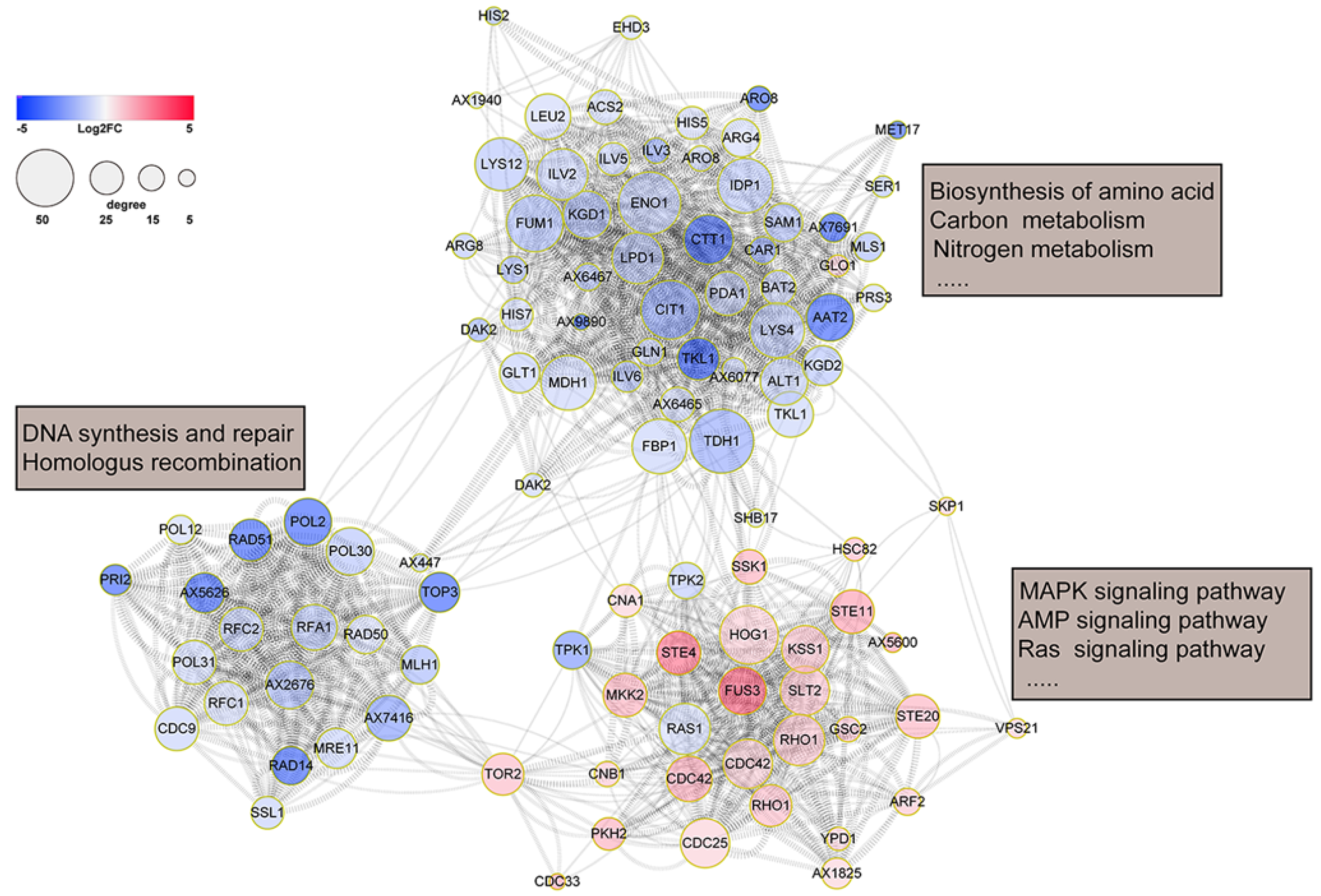
3.4. MAPK Signaling Pathway in T. fuciformis
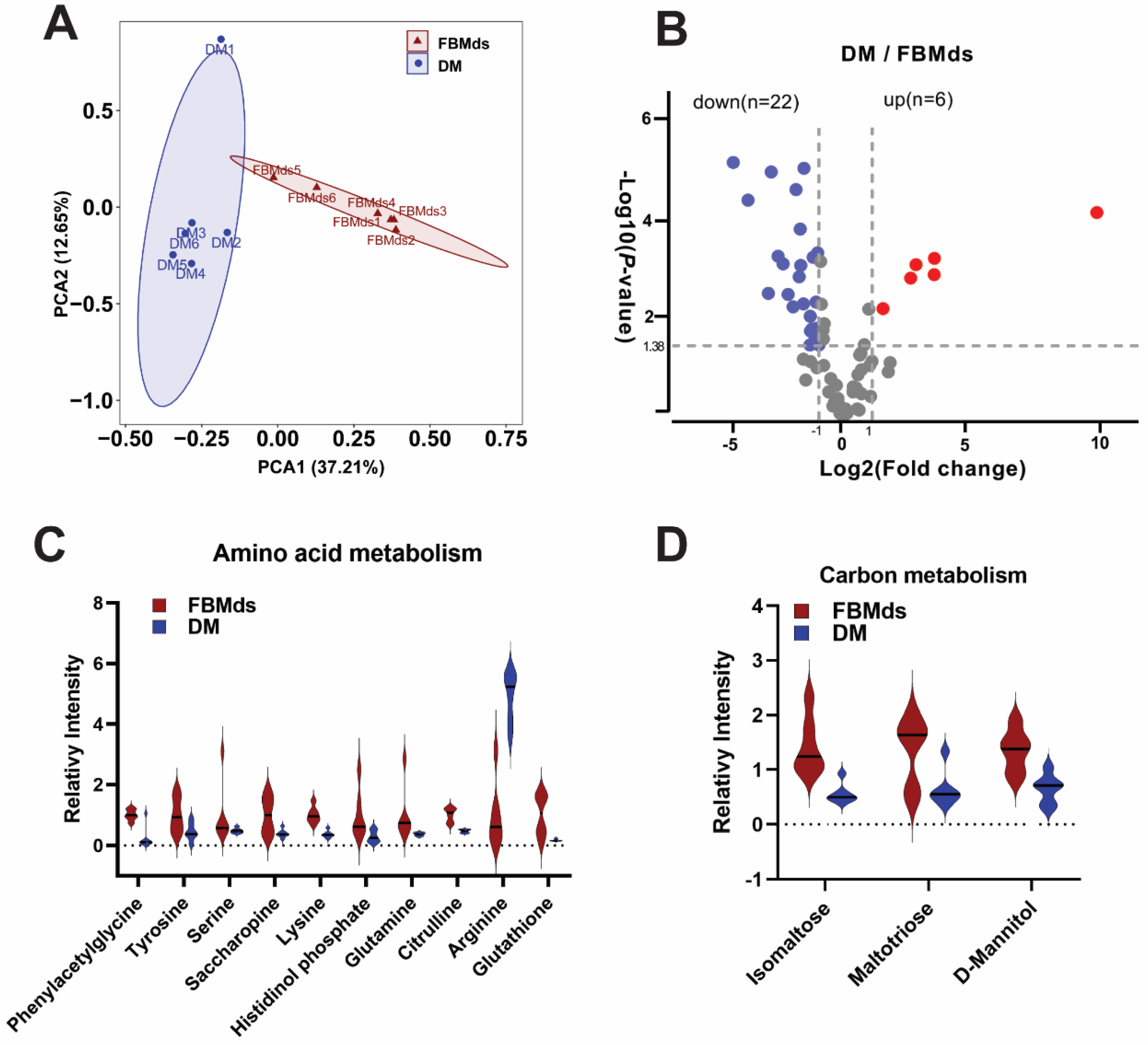
3.5. Comparative Metabolism of FBMds and DM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FBMds | Dikaryotic yeast-like conidia from fruiting bodies and mycelia |
| DM | Dikaryotic mycelia |
| DIA | Data-independent acquisition |
| HRMS1-DIA | high-resolution MS1-based quantitative data-independent acquisition |
| PRM | Parallel reaction monitoring |
| DEPs | Differentially expressed proteins |
| GO | Gene ontology |
| KEGG | Kyoto encyclopedia of genes and genomes |
| MAPK | Mitogen-activated protein kinase |
| TPS | Tremella polysaccharide |
| cAMP–PKA | cAMP–protein kinase A |
| TOR | Target of rapamycin |
| DDA | Data-dependent acquisition |
| SWATH-MS | Sequential window acquisition of all theoretical mass spectra |
| PDA | Potato dextrose agar |
| FDR | False discovery rate |
| BP | Biological process |
| MF | Molecular function |
| CC | Cellular component |
| PCA | Principal components analysis |
References
- Liu, J.; Ma, A.M.; Sheng, G.H.; Chen, L.G. Preliminary study on the differences of dimorphic cells in Tremella fuciformis. Microbiol. China 2007, 30, 53–57. [Google Scholar]
- Chen, L.D.; Liu, Y.C.; Kong, X.Q.; Zhang, Q.H. Conditions for germination of dikaryotic yeast-like spores from fruiting body and mycelium of Tremella fuciformis. Edible Fungi China 2021, 40, 12–18. [Google Scholar]
- Wu, Y.J.; Wei, Z.X.; Zhang, F.M.; Linhardt, R.J.; Sun, P.L.; Zhang, A.Q. Structure, bioactivities and applications of the polysaccharides from Tremella fuciformis mushroom: A review. Int. J. Biol. Macromol. 2019, 121, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Iturbe-Espinoza, P.; Gil-Moreno, S.; Lin, W.Y.; Calatayud, S.; Palacios, O.; Capdevila, M.; Atrian, S. The fungus Tremella mesenterica encodes the longest metallothionein currently known: Gene, protein and metal binding characterization. PLoS ONE 2016, 11, e0148651. [Google Scholar]
- Zhang, B.Z.; Zhu, L.Y.; Zhang, W.D. Study on the morphology of Tremella fuciformis mycelium and the preparation of Tremella fuciformis mother species. Edible Fungi China 1995, 14, 17–20. [Google Scholar]
- Zhou, Y.L.; Fang, G. Preliminary study on nutritional conditions of germinationy of Tremella fuciformis. Edible Fungi China 1985, 32, 4–6. [Google Scholar]
- Liu, J.; Wu, Y.; Ma, A.M.; Chen, L.G. Research progress of environmental factors and signal transduction pathways of dimorphism in fungi. Microbiol. China 2008, 35, 1102–1106. [Google Scholar]
- Zhu, H.Y.; Yuan, Y.; Liu, J.; Zheng, L.S.; Chen, L.G.; Ma, A.M. Comparing the sugar profiles and primary structures of alkali-extracted water-soluble polysaccharides in cell wall between the yeast and mycelial phases from Tremella fuciformis. J. Microbiol. 2016, 54, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.Y.; Liu, D.M.; Zheng, L.S.; Chen, L.G.; Ma, A.M. Characterization of a G protein N1 subunit encoded gene from the dimorphic fungus-Tremella fuciformis. Antonie Leeuwenhoek 2021, 114, 1949–1960. [Google Scholar] [CrossRef]
- Boyce, K.J.; Andrianopoulos, A. Fungal dimorphism: The switch from hyphae to yeast is a specialized morphogenetic adaptation allowing colonization of a host. FEMS Microbiol. Rev. 2015, 39, 797–811. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, A.; Oviedo, C.; Nickerson, K.W.; Navarrete, J. Quorum sensing activity and control of yeast-mycelium dimorphism in Ophiostoma floccosum. Biotechnol. Lett. 2014, 36, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Q.; Tian, X.Y.; Gyawali, R.; Lin, X.R. Fungal adhesion protein guides community behaviors and autoinduction in a paracrine manner. Proc. Natl. Acad. Sci. USA 2013, 110, 11571–11576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Tian, X.; Gyawali, R.; Upadhyay, S.; Foyle, D.; Wang, G.; Cai, J.J.; Lin, X. Morphotype transition and sexual reproduction are genetically associated in a ubiquitous environmental pathogen. PLoS Pathog. 2014, 10, e1004185. [Google Scholar] [CrossRef] [PubMed]
- Barnouin, K. Two-dimensional gel electrophoresis for analysis of protein complexes. Methods Mol. Biol. 2004, 261, 479–498. [Google Scholar] [PubMed]
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031. [Google Scholar] [CrossRef] [Green Version]
- Vidova, V.; Spacil, Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Anal. Chim. Acta 2017, 964, 7–23. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef] [Green Version]
- Xuan, Y.; Bateman, N.W.; Gallien, S.; Goetze, S.; Zhou, Y.; Navarro, P.; Hu, M.; Parikh, N.; Hood, B.L.; Conrads, K.A.; et al. Standardization and harmonization of distributed multi-center proteotype analysis supporting precision medicine studies. Nat. Commun. 2020, 11, 5248. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Yang, P.; Li, Y.; He, C.; Yan, J.; Zhang, W.; Li, X.; Xiang, F.; Zuo, Z.; Li, X.; Zhu, Y.; et al. Phenotype and TMT-based quantitative proteomics analysis of Brassica napus reveals new insight into chlorophyll synthesis and chloroplast structure. J. Proteom. 2020, 214, 103621. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Xue, J.; Balamurugan, S.; Li, D.W.; Liu, Y.H.; Zeng, H.; Wang, L.; Yang, W.D.; Liu, J.S.; Li, H.Y. Glucose-6-phosphate dehydrogenase as a target for highly efficient fatty acid biosynthesis in microalgae by enhancing NADPH supply. Metab. Eng. 2017, 41, 212–221. [Google Scholar] [CrossRef]
- Li, H.; Cai, Y.; Guo, Y.; Chen, F.; Zhu, Z.J. MetDIA: Targeted metabolite extraction of multiplexed MS/MS spectra generated by data-Independent acquisition. Anal. Chem. 2016, 88, 8757–8764. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A web-based platform to process untargeted metabolomic data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Lopez, C.; Collette, J.R.; Brothers, K.M.; Shepardson, K.M.; Cramer, R.A.; Wheeler, R.T.; Lorenz, M.C. Candida albicans induces arginine biosynthetic genes in response to host-derived reactive oxygen species. Eukaryot. Cell 2013, 12, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.F.; Cao, Q.C.; Hu, P.; Cui, H.F.; Yu, X.P.; Ye, Z.H. Investigation on the differentiation of two Ustilago esculenta strains—Implications of a relationship with the host phenotypes appearing in the fields. BMC Microbiol. 2017, 17, 228. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Navarathna, D.H.; Roberts, D.D.; Cooper, J.T.; Atkin, A.L.; Petro, T.M.; Nickerson, K.W. Arginine-induced germ tube formation in Candida albicans is essential for escape from murine macrophage line RAW 264.7. Infect. Immun. 2009, 77, 1596–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, D.S.; Pereira, M.; Portis, I.G.; Dos Santos Junior, A.C.M.; Fontes, W.; de Sousa, M.V.; Assuncao, L.D.P.; Baeza, L.C.; Bailao, A.M.; Ricart, C.A.O.; et al. Metabolic peculiarities of Paracoccidioides brasiliensis dimorphism as demonstrated by iTRAQ labeling proteomics. Front. Microbiol. 2019, 10, 555. [Google Scholar] [CrossRef]
- Tamayo, D.; Muñoz, J.F.; Lopez, Á.; Urán, M.; Herrera, J.; Borges, C.L.; Restrepo, Á.; Soares, C.M.; Taborda, C.P.; Almeida, A.J.; et al. Identification and analysis of the role of superoxide dismutases isoforms in the pathogenesis of Paracoccidioides spp. PLoS Negl. Trop. Dis. 2016, 10, e0004481. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Tsang, C.C.; Woo, P.C.Y. Talaromyces marneffei genomic, transcriptomic, proteomic and metabolomic studies reveal mechanisms for environmental adaptations and virulence. Toxins 2017, 9, 192. [Google Scholar] [CrossRef] [Green Version]
- Shaykholeslam Esfahani, E.; Shahpiri, A. Thioredoxin h isoforms from rice are differentially reduced by NADPH/thioredoxin or GSH/glutaredoxin systems. Int. J. Biol. Macromol. 2015, 74, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.T.; Ahn, C.S.; Kim, J.G.; Ro, H.S.; Lee, C.W.; Kim, J.W. Proteomic analysis of early phase of conidia germination in Aspergillus nidulans. Fungal Genet. Biol. 2010, 47, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Osherov, N.; May, G. Conidial germination in Aspergillus nidulans requires RAS signaling and protein synthesis. Genetics 2000, 155, 647–656. [Google Scholar] [CrossRef]
- Chen, R.E.; Thorner, J. Function and regulation in MAPK signaling pathways: Lessons learned from the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2007, 1773, 1311–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozaki, S.; Furuya, K.; Niki, H. The Ras1-Cdc42 pathway is involved in hyphal development of Schizosaccharomyces japonicus. FEMS Yeast Res. 2018, 18, foy031. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Mehrabi, R.; Xu, J.R. Mitogen-activated protein kinase pathways and fungal pathogenesis. Eukaryot. Cell 2007, 6, 1701–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, P.; Aichinger, C.; Feldbrügge, M.; Kahmann, R. The MAP kinase kpp2 regulates mating and pathogenic development in Ustilago maydis. Mol. Microbiol. 1999, 34, 1007–1017. [Google Scholar] [CrossRef]
- Müller, P.; Weinzierl, G.; Brachmann, A.; Feldbrügge, M.; Kahmann, R. Mating and pathogenic development of the Smut fungus Ustilago maydis are regulated by one mitogen-activated protein kinase cascade. Eukaryot. Cell 2003, 2, 1187–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Heitman, J. Cyclic AMP-dependent protein kinase regulates pseudohyphal differentiation in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999, 19, 4874–4887. [Google Scholar] [CrossRef] [Green Version]
- Karunanithi, S.; Cullen, P.J. The filamentous growth MAPK pathway responds to glucose starvation through the Mig1/2 transcriptional repressors in Saccharomyces cerevisiae. Genetics 2012, 192, 869–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conceicao, P.M.; Chaves, A.F.A.; Navarro, M.V.; Castilho, D.G.; Calado, J.C.P.; Haniu, A.; Xander, P.; Batista, W.L. Cross-talk between the Ras GTPase and the Hog1 survival pathways in response to nitrosative stress in Paracoccidioides brasiliensis. Nitric Oxide 2019, 86, 1–11. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Tang, H.; Zhao, W.; Yang, Y.; Fan, X.; Zhan, G.; Li, J.; Sun, S. Study of Dimorphism Transition Mechanism of Tremella fuciformis Based on Comparative Proteomics. J. Fungi 2022, 8, 242. https://doi.org/10.3390/jof8030242
Li Y, Tang H, Zhao W, Yang Y, Fan X, Zhan G, Li J, Sun S. Study of Dimorphism Transition Mechanism of Tremella fuciformis Based on Comparative Proteomics. Journal of Fungi. 2022; 8(3):242. https://doi.org/10.3390/jof8030242
Chicago/Turabian StyleLi, Yaxing, Haohao Tang, Weichao Zhao, Yang Yang, Xiaolu Fan, Guanping Zhan, Jiahuan Li, and Shujing Sun. 2022. "Study of Dimorphism Transition Mechanism of Tremella fuciformis Based on Comparative Proteomics" Journal of Fungi 8, no. 3: 242. https://doi.org/10.3390/jof8030242
APA StyleLi, Y., Tang, H., Zhao, W., Yang, Y., Fan, X., Zhan, G., Li, J., & Sun, S. (2022). Study of Dimorphism Transition Mechanism of Tremella fuciformis Based on Comparative Proteomics. Journal of Fungi, 8(3), 242. https://doi.org/10.3390/jof8030242