
Fun30 and Rtt109 Mediate Epigenetic Regulation of the DNA Damage Response Pathway in C. albicans

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Plasmids and Strains
2.3. Antibodies
2.4. Generation of C. albicans and S. cerevisiae Mutant Strains
2.5. Generation of Overexpression Strains
2.6. Generation of Revertant Strains
2.7. Epitope-Tagging of Endogenous FUN30
2.8. Cloning of C. albicans FUN30 into a Mammalian Expression Vector
2.9. Functional Complementation Assays
2.10. DNA End Resection
2.11. Quantitative PCR (qPCR)
2.12. Chromatin Immunoprecipitation (ChIP) Assay
2.13. Western Blots
2.14. Immunofluorescence
2.15. Growth Rate Analysis
2.16. Plate Assays
2.17. Minimal Inhibitory Concentration 50 (MIC50)
2.18. Phylogenetic and Domain Analysis
2.19. Statistical Analysis
3. Results
3.1. The orf19.6291 Encodes Fun30, a Member of the ATP-Dependent Chromatin Remodeling Protein Family
3.2. Fun30 Localizes to the Nucleus
3.3. C. albicans Fun30 Is a Functional Homolog of Both S. cerevisiae Fun30 and Human SMARCAD1
3.4. Fun30 Protein of C. albicans Mediates Double-Strand Break End Resection
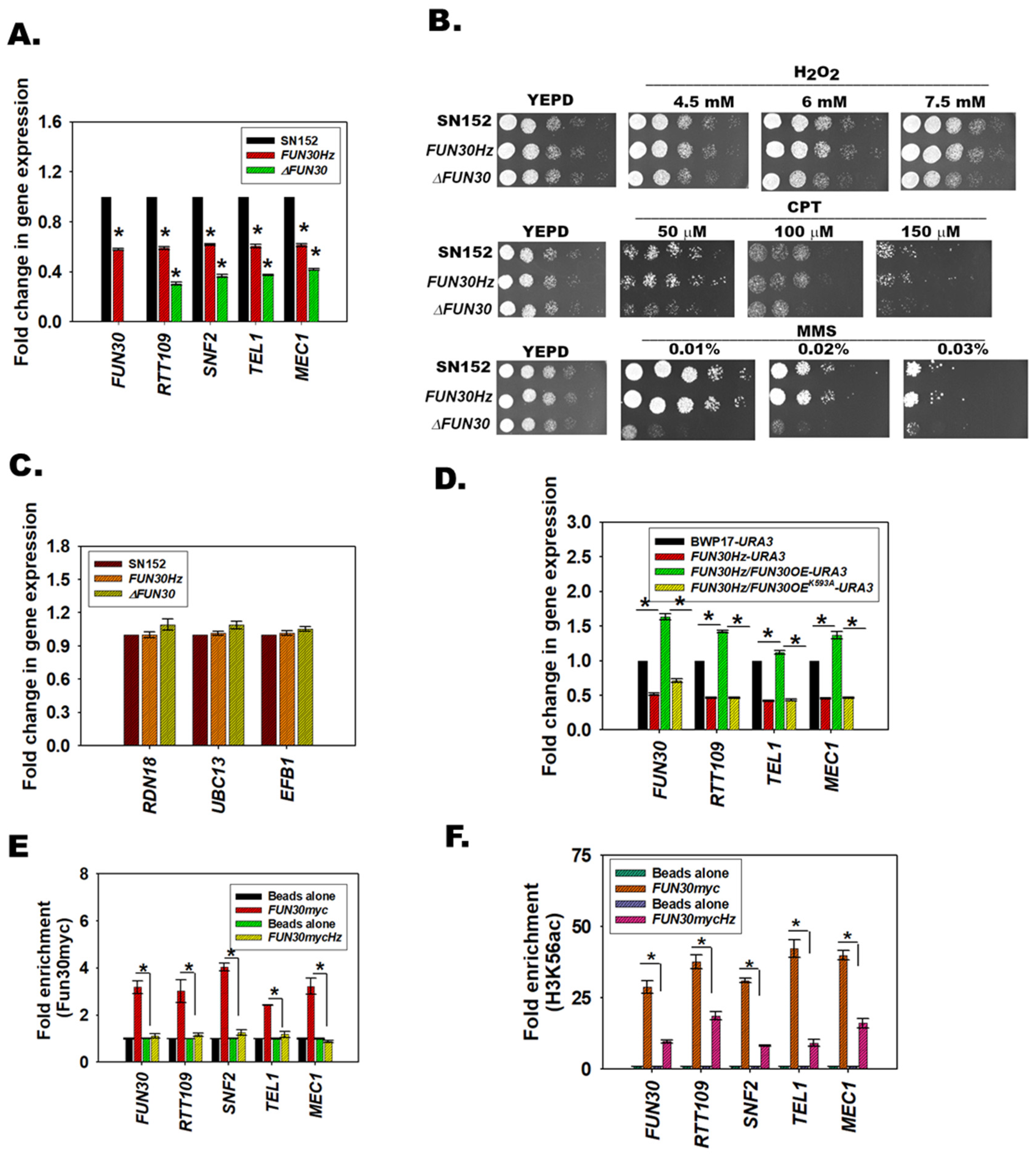
3.5. FUN30 Mediates the Response to Genotoxic Stress
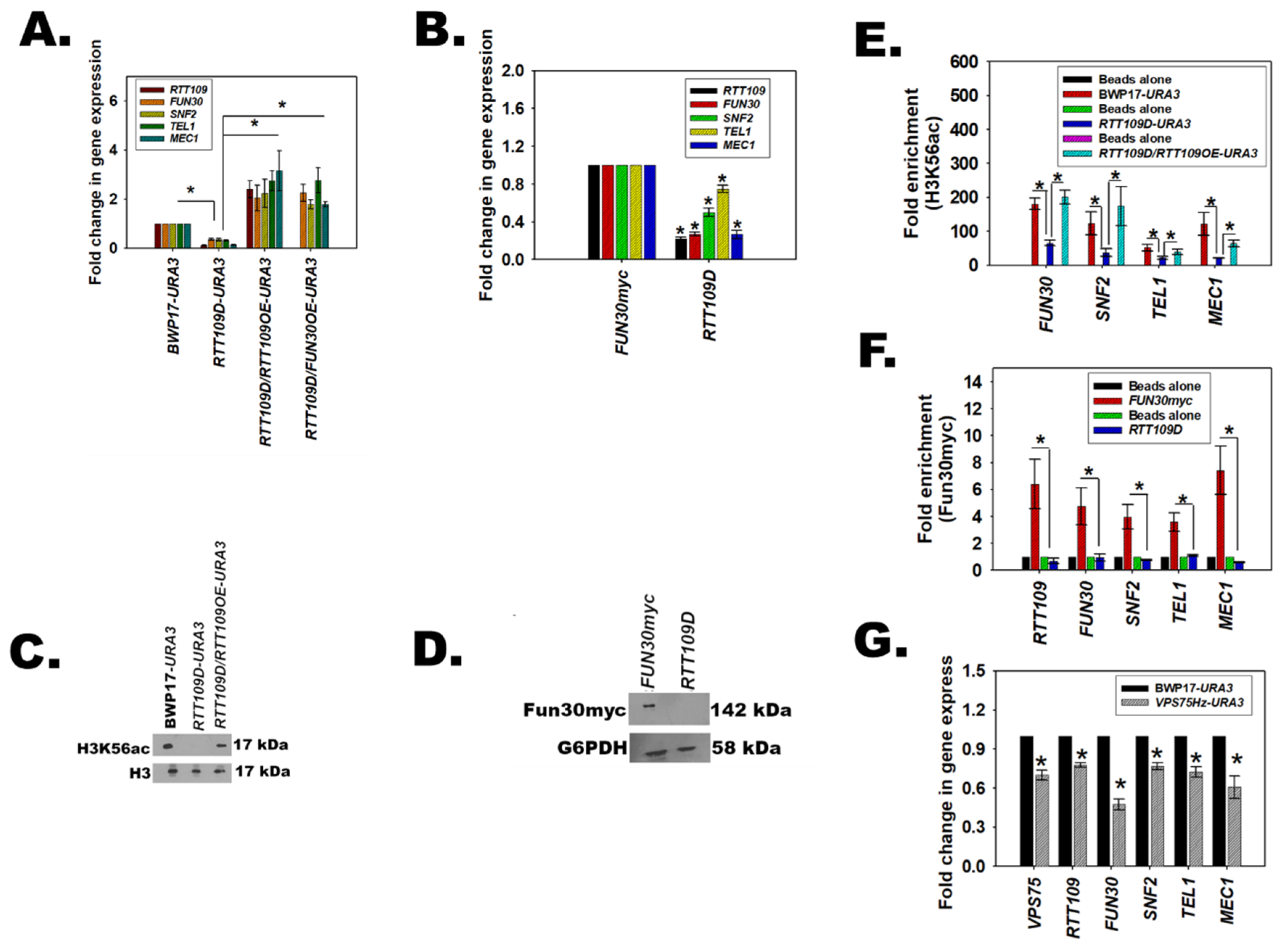
3.6. Fun30 Regulates the Expression of RTT109, SNF2, TEL1, and MEC1 in C. albicans
3.7. The ATPase Activity of Fun30 Is Needed for Transcriptional Regulation
3.8. Fun30 Binds to the Promoter Regions of the DNA Damage Response Genes
3.9. H3K56ac Regulates the Expression of FUN30, SNF2, TEL1, and MEC1
3.10. The Catalytic Activity of Rtt109 Is Essential for the Transcription Regulation
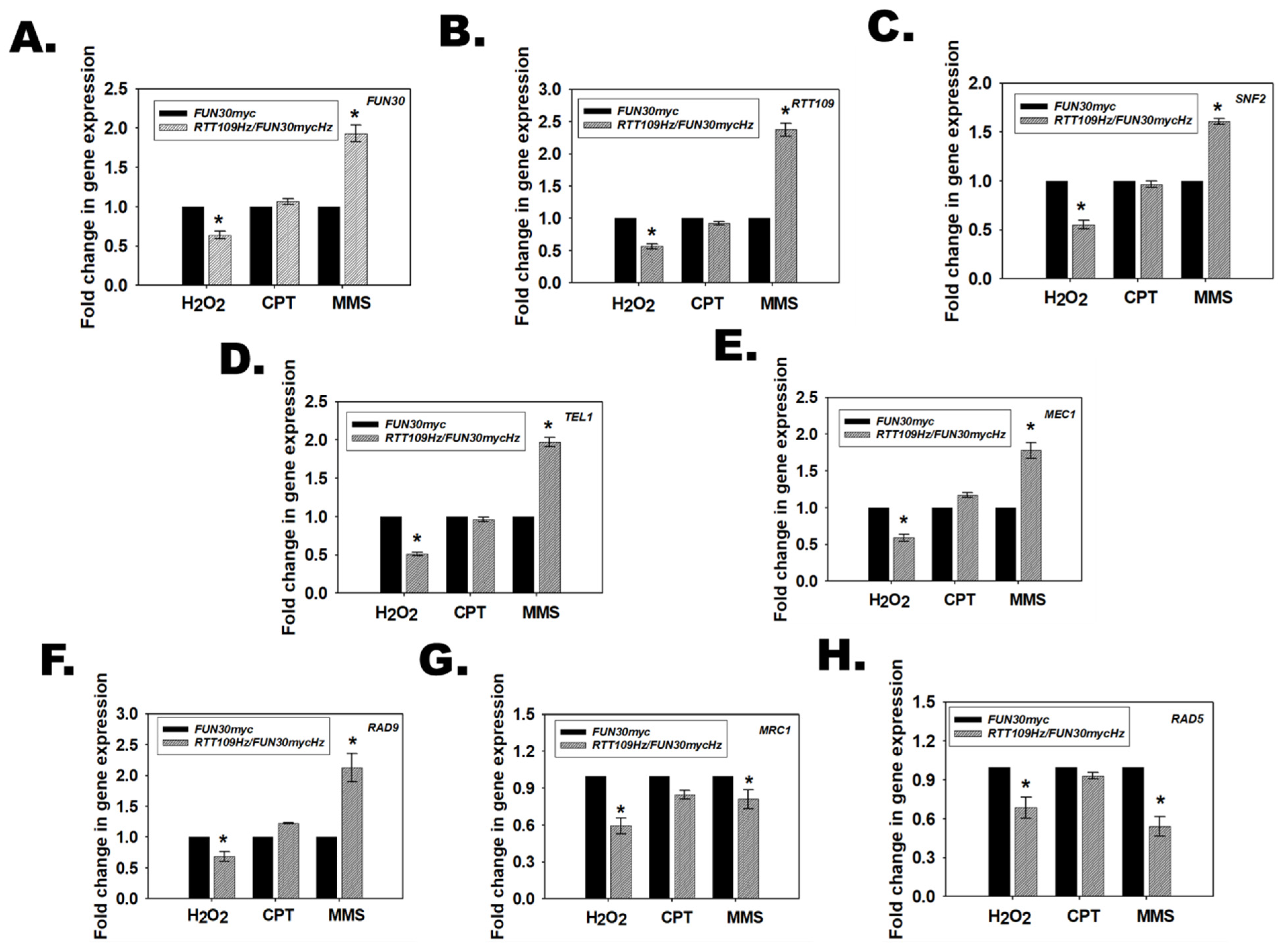
3.11. RTT109Hz/FUN30Hz Double Mutant Shows a Differential Response to Genotoxic Stress
3.12. Rtt109 and Fun30 in RTT109Hz/FUN30Hz Regulate the Transcription of DNA Damage Response Pathways Genes Differentially in Response to Different Types of Genotoxic Stressors
3.13. The Response of RTT109Hz/FUN30Hz to DNA Damage Is Regulated by the Occupancy of Fun30 and H3K56ac on RAD9, MRC1, and RAD5 Promoter
3.14. Single Copy Deletion of RAD9 in RTT109Hz/FUN30Hz Rescues the Resistance to MMS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morettini, S.; Podhraski, V.; Lusser, A. ATP-Dependent Chromatin Remodeling Enzymes and Their Various Roles in Cell Cycle Control. Front. Biosci. J. Virtual Libr. 2008, 13, 5522–5532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaus, A.; Martin, D.M.A.; Barton, G.J.; Owen-Hughes, T. Identification of Multiple Distinct Snf2 Subfamilies with Conserved Structural Motifs. Nucleic Acids Res. 2006, 34, 2887–2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbalenya, A.E.; Koonin, E.V. Helicases: Amino Acid Sequence Comparisons and Structure-Function Relationships. Curr. Opin. Struct. Biol. 1993, 3, 419–429. [Google Scholar] [CrossRef]
- Flaus, A.; Owen-Hughes, T. Mechanisms for ATP-Dependent Chromatin Remodelling: The Means to the End. FEBS J. 2011, 278, 3579–3595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuguchi, G.; Shen, X.; Landry, J.; Wu, W.-H.; Sen, S.; Wu, C. ATP-Driven Exchange of Histone H2AZ Variant Catalyzed by SWR1 Chromatin Remodeling Complex. Science 2004, 303, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Eapen, V.V.; Sugawara, N.; Tsabar, M.; Wu, W.-H.; Haber, J.E. The Saccharomyces Cerevisiae Chromatin Remodeler Fun30 Regulates DNA End Resection and Checkpoint Deactivation. Mol. Cell. Biol. 2012, 32, 4727–4740. [Google Scholar] [CrossRef] [Green Version]
- Costelloe, T.; Louge, R.; Tomimatsu, N.; Mukherjee, B.; Martini, E.; Khadaroo, B.; Dubois, K.; Wiegant, W.W.; Thierry, A.; Burma, S.; et al. The Yeast Fun30 and Human SMARCAD1 Chromatin Remodellers Promote DNA End Resection. Nature 2012, 489, 581–584. [Google Scholar] [CrossRef] [Green Version]
- Neves-Costa, A.; Will, W.R.; Vetter, A.T.; Miller, J.R.; Varga-Weisz, P. The SNF2-Family Member Fun30 Promotes Gene Silencing in Heterochromatic Loci. PLoS ONE 2009, 4, e8111. [Google Scholar] [CrossRef] [Green Version]
- Steglich, B.; Strålfors, A.; Khorosjutina, O.; Persson, J.; Smialowska, A.; Javerzat, J.-P.; Ekwall, K. The Fun30 Chromatin Remodeler Fft3 Controls Nuclear Organization and Chromatin Structure of Insulators and Subtelomeres in Fission Yeast. PLoS Genet. 2015, 11, e1005101. [Google Scholar] [CrossRef] [Green Version]
- Awad, S.; Ryan, D.; Prochasson, P.; Owen-Hughes, T.; Hassan, A.H. The Snf2 Homolog Fun30 Acts as a Homodimeric ATP-Dependent Chromatin-Remodeling Enzyme. J. Biol. Chem. 2010, 285, 9477–9484. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.; Bajwa, P.; Johnson, F.C.; Bhaumik, S.R.; Shilatifard, A. Rtt109 Is Required for Proper H3K56 Acetylation A Chromatin Mark Associated with the Elongating RNA Polymerase II. J. Biol. Chem. 2006, 281, 37270–37274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arcy, S.; Luger, K. Understanding Histone Acetyltransferase Rtt109 Structure and Function: How Many Chaperones Does It Take? Curr. Opin. Struct. Biol. 2011, 21, 728–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driscoll, R.; Hudson, A.; Jackson, S.P. Yeast Rtt109 Promotes Genome Stability by Acetylating Histone H3 on Lysine 56. Science 2007, 315, 649–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, J.L.d.; Boyartchuk, V.L.; Zhu, L.J.; Kaufman, P.D. Histone Acetyltransferase Rtt109 Is Required for Candida Albicans Pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 1594–1599. [Google Scholar] [CrossRef] [Green Version]
- Vempati, R.K.; Jayani, R.S.; Notani, D.; Sengupta, A.; Galande, S.; Haldar, D. P300-Mediated Acetylation of Histone H3 Lysine 56 Functions in DNA Damage Response in Mammals. J. Biol. Chem. 2010, 285, 28553–28564. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Holbert, M.A.; Wurtele, H.; Meeth, K.; Rocha, W.; Gharib, M.; Jiang, E.; Thibault, P.; Verreault, A.; Cole, P.A.; et al. Fungal Rtt109 Histone Acetyltransferase Is an Unexpected Structural Homolog of Metazoan P300/CBP. Nat. Struct. Mol. Biol. 2008, 15, 738–745. [Google Scholar] [CrossRef]
- Jain, P.; Garai, P.; Sethi, S.C.; Naqvi, N.; Yadav, B.; Kumar, P.; Singh, S.L.; Yadav, U.; Bhatnagar, S.; Rahul; et al. Modulation of Azole Sensitivity and Filamentation by GPI15, Encoding a Subunit of the First GPI Biosynthetic Enzyme, in Candida Albicans. Sci. Rep. 2019, 9, 8508. [Google Scholar] [CrossRef]
- Dantas, A.; Day, A.; Ikeh, M.; Kos, I.; Achan, B.; Quinn, J. Oxidative Stress Responses in the Human Fungal Pathogen, Candida Albicans. Biomolecules 2015, 5, 142–165. [Google Scholar] [CrossRef] [Green Version]
- Walther, A.; Wendland, J. An Improved Transformation Protocol for the Human Fungal Pathogen Candida Albicans. Curr. Genet. 2003, 42, 339–343. [Google Scholar] [CrossRef]
- Bi, X.; Yu, Q.; Siler, J.; Li, C.; Khan, A. Functions of Fun30 Chromatin Remodeler in Regulating Cellular Resistance to Genotoxic Stress. PLoS ONE 2015, 10, e0121341. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods San Diego Calif. 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Singh-Babak, S.D.; Babak, T.; Diezmann, S.; Hill, J.A.; Xie, J.L.; Chen, Y.-L.; Poutanen, S.M.; Rennie, R.P.; Heitman, J.; Cowen, L.E. Global Analysis of the Evolution and Mechanism of Echinocandin Resistance in Candida Glabrata. PLoS Pathog. 2012, 8, e1002718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inglis, D.O.; Arnaud, M.B.; Binkley, J.; Shah, P.; Skrzypek, M.S.; Wymore, F.; Binkley, G.; Miyasato, S.R.; Simison, M.; Sherlock, G. The Candida Genome Database Incorporates Multiple Candida Species: Multispecies Search and Analysis Tools with Curated Gene and Protein Information for Candida Albicans and Candida Glabrata. Nucleic Acids Res. 2012, 40, D667–D674. [Google Scholar] [CrossRef] [PubMed]
- Shih, S.C.; Prag, G.; Francis, S.A.; Sutanto, M.A.; Hurley, J.H.; Hicke, L. A Ubiquitin-Binding Motif Required for Intramolecular Monoubiquitylation, the CUE Domain. EMBO J. 2003, 22, 1273–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Pandita, R.K.; Hambarde, S.; Mattoo, A.R.; Charaka, V.; Ahmed, K.M.; Iyer, S.P.; Hunt, C.R.; Pandita, T.K. SMARCAD1 Phosphorylation and Ubiquitination Are Required for Resection during DNA Double-Strand Break Repair. iScience 2018, 2, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byeon, B.; Wang, W.; Barski, A.; Ranallo, R.T.; Bao, K.; Schones, D.E.; Zhao, K.; Wu, C.; Wu, W.-H. The ATP-Dependent Chromatin Remodeling Enzyme Fun30 Represses Transcription by Sliding Promoter-Proximal Nucleosomes. J. Biol. Chem. 2013, 288, 23182–23193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Choi, E.S.; Seo, H.D.; Kang, K.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Choe, J.; Workman, J.L.; Lee, D. Chromatin Remodeller Fun30Fft3 Induces Nucleosome Disassembly to Facilitate RNA Polymerase II Elongation. Nat. Commun. 2017, 8, 14527. [Google Scholar] [CrossRef]
- Lee, J.; Choi, E.S.; Lee, D. It’s Fun to Transcribe with Fun30: A Model for Nucleosome Dynamics during RNA Polymerase II-Mediated Elongation. Transcription 2018, 9, 108–116. [Google Scholar] [CrossRef]
- Sethy, R.; Rakesh, R.; Patne, K.; Arya, V.; Sharma, T.; Haokip, D.T.; Kumari, R.; Muthuswami, R. Regulation of ATM and ATR by SMARCAL1 and BRG1. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 1076–1092. [Google Scholar] [CrossRef] [Green Version]
- Patne, K.; Rakesh, R.; Arya, V.; Chanana, U.B.; Sethy, R.; Swer, P.B.; Muthuswami, R. BRG1 and SMARCAL1 Transcriptionally Co-Regulate DROSHA, DGCR8 and DICER in Response to Doxorubicin-Induced DNA Damage. Biochim. Biophys. Acta 2017, 1860, 936–951. [Google Scholar] [CrossRef]
- Haokip, D.T.; Goel, I.; Arya, V.; Sharma, T.; Kumari, R.; Priya, R.; Singh, M.; Muthuswami, R. Transcriptional Regulation of ATP-Dependent Chromatin Remodeling Factors: SMARCAL1 and BRG1 Mutually Co-Regulate Each Other. Sci. Rep. 2016, 6, 20532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craven, R.J.; Greenwell, P.W.; Dominska, M.; Petes, T.D. Regulation of Genome Stability by TEL1 and MEC1, Yeast Homologs of the Mammalian ATM and ATR Genes. Genetics 2002, 161, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Park, J.-H.; Kim, S.-J.; Kwon, S.-J.; Kwon, J. A Cooperative Activation Loop among SWI/SNF, γ-H2AX and H3 Acetylation for DNA Double-Strand Break Repair. EMBO J. 2010, 29, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.-J.; Park, J.-H.; Park, E.-J.; Lee, S.-A.; Lee, H.-S.; Kang, S.W.; Kwon, J. ATM-Mediated Phosphorylation of the Chromatin Remodeling Enzyme BRG1 Modulates DNA Double-Strand Break Repair. Oncogene 2015, 34, 303–313. [Google Scholar] [CrossRef]
- Tsubota, T.; Berndsen, C.E.; Erkmann, J.A.; Smith, C.L.; Yang, L.; Freitas, M.A.; Denu, J.M.; Kaufman, P.D. Histone H3-K56 Acetylation Is Catalyzed by Histone Chaperone-Dependent Complexes. Mol. Cell 2007, 25, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Stojic, L.; Brun, R.; Jiricny, J. Mismatch Repair and DNA Damage Signalling. DNA Repair 2004, 3, 1091–1101. [Google Scholar] [CrossRef]
- Henle, E.S.; Linn, S. Formation, Prevention, and Repair of DNA Damage by Iron/Hydrogen Peroxide. J. Biol. Chem. 1997, 272, 19095–19098. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y. Topoisomerase I Inhibitors: Camptothecins and Beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef]
- Osborn, A.J. Mrc1 Is a Replication Fork Component Whose Phosphorylation in Response to DNA Replication Stress Activates Rad53. Genes Dev. 2003, 17, 1755–1767. [Google Scholar] [CrossRef] [Green Version]
- Unk, I.; Hajdú, I.; Blastyák, A.; Haracska, L. Role of Yeast Rad5 and Its Human Orthologs, HLTF and SHPRH in DNA Damage Tolerance. DNA Repair 2010, 9, 257–267. [Google Scholar] [CrossRef]
- Alcasabas, A.A.; Osborn, A.J.; Bachant, J.; Hu, F.; Werler, P.J.; Bousset, K.; Furuya, K.; Diffley, J.F.; Carr, A.M.; Elledge, S.J. Mrc1 Transduces Signals of DNA Replication Stress to Activate Rad53. Nat. Cell Biol. 2001, 3, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.F.; Duong, J.K.; Sun, Z.; Morrow, J.S.; Pradhan, D.; Stern, D.F. Rad9 Phosphorylation Sites Couple Rad53 to the Saccharomyces Cerevisiae DNA Damage Checkpoint. Mol. Cell 2002, 9, 1055–1065. [Google Scholar] [CrossRef]
- Allen, J.B.; Zhou, Z.; Siede, W.; Friedberg, E.C.; Elledge, S.J. The SAD1/RAD53 Protein Kinase Controls Multiple Checkpoints and DNA Damage-Induced Transcription in Yeast. Genes Dev. 1994, 8, 2401–2415. [Google Scholar] [CrossRef] [Green Version]
- Neecke, H. Cell Cycle Progression in the Presence of Irreparable DNA Damage Is Controlled by a Mec1- and Rad53-Dependent Checkpoint in Budding Yeast. EMBO J. 1999, 18, 4485–4497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinert, T.; Hartwell, L. Control of G2 Delay by the Rad9 Gene of Saccharomyces Cerevisiae. J. Cell Sci. Suppl. 1989, 12, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Aboussekhra, A.; Vialard, J.E.; Morrison, D.E.; Torre-Ruiz, M.A.d.l.; Cernáková, L.; Fabre, F.; Lowndes, N.F. A Novel Role for the Budding Yeast RAD9 Checkpoint Gene in DNA Damage-Dependent Transcription. EMBO J. 1996, 15, 3912–3922. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Bergmaier, P.; Sachs, P.; Klangwart, M.; Rückert, T.; Bartels, N.; Demmers, J.; Dekker, M.; Poot, R.A.; Mermoud, J.E. The CUE1 Domain of the SNF2-like Chromatin Remodeler SMARCAD1 Mediates Its Association with KRAB-Associated Protein 1 (KAP1) and KAP1 Target Genes. J. Biol. Chem. 2018, 293, 2711–2724. [Google Scholar] [CrossRef] [Green Version]
- Durand-Dubief, M.; Will, W.R.; Petrini, E.; Theodorou, D.; Harris, R.R.; Crawford, M.R.; Paszkiewicz, K.; Krueger, F.; Correra, R.M.; Vetter, A.T.; et al. SWI/SNF-Like Chromatin Remodeling Factor Fun30 Supports Point Centromere Function in S. Cerevisiae. PLoS Genet. 2012, 8, e1002974. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Bazán, M.Á.; Gallo-Fernández, M.; Saugar, I.; Jiménez-Martín, A.; Vázquez, M.V.; Tercero, J.A. Rad5 Plays a Major Role in the Cellular Response to DNA Damage during Chromosome Replication. Cell Rep. 2014, 9, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Cui, D.; Papusha, A.; Zhang, X.; Chu, C.-D.; Tang, J.; Chen, K.; Pan, X.; Ira, G. The Fun30 Nucleosome Remodeller Promotes Resection of DNA Double-Strand Break Ends. Nature 2012, 489, 576–580. [Google Scholar] [CrossRef] [Green Version]
- Christmann, M.; Kaina, B. Epigenetic Regulation of DNA Repair Genes and Implications for Tumor Therapy. Mutat. Res. 2019, 780, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Raab, J.R.; Resnick, S.; Magnuson, T. Genome-Wide Transcriptional Regulation Mediated by Biochemically Distinct SWI/SNF Complexes. PLoS Genet. 2015, 11, e1005748. [Google Scholar] [CrossRef] [PubMed]
- Victoria, G.S.; Kumar, P.; Komath, S.S. The Candida Albicans Homologue of PIG-P, CaGpi19p: Gene Dosage and Role in Growth and Filamentation. Microbiology 2010, 156, 3041–3051. [Google Scholar] [CrossRef]
- Wilson, R.B.; Davis, D.; Mitchell, A.P. Rapid Hypothesis Testing with Candida Albicans through Gene Disruption with Short Homology Regions. J. Bacteriol. 1999, 181, 1868–1874. [Google Scholar] [CrossRef] [Green Version]
- Gerami-Nejad, M.; Hausauer, D.; McClellan, M.; Berman, J.; Gale, C. Cassettes for the PCR-Mediated Construction of Regulatable Alleles in Candida Albicans. Yeast 2004, 21, 429–436. [Google Scholar] [CrossRef]
- Noble, S.M.; Johnson, A.D. Strains and Strategies for Large-Scale Gene Deletion Studies of the Diploid Human Fungal Pathogen Candida Albicans. Eukaryot. Cell 2005, 4, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Sikorski, R.S.; Hieter, P. A System of Shuttle Vectors and Yeast Host Strains Designed for Efficient Manipulation of DNA in Saccharomyces Cerevisiae. Genetics 1989, 122, 19–27. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maurya, P.K.; Garai, P.; Goel, K.; Bhatt, H.; Dutta, A.; Goyal, A.; Dewasthale, S.; Gupta, M.; Haokip, D.T.; Barik, S.; et al. Fun30 and Rtt109 Mediate Epigenetic Regulation of the DNA Damage Response Pathway in C. albicans. J. Fungi 2022, 8, 559. https://doi.org/10.3390/jof8060559
Maurya PK, Garai P, Goel K, Bhatt H, Dutta A, Goyal A, Dewasthale S, Gupta M, Haokip DT, Barik S, et al. Fun30 and Rtt109 Mediate Epigenetic Regulation of the DNA Damage Response Pathway in C. albicans. Journal of Fungi. 2022; 8(6):559. https://doi.org/10.3390/jof8060559
Chicago/Turabian StyleMaurya, Prashant Kumar, Pramita Garai, Kaveri Goel, Himanshu Bhatt, Anindita Dutta, Aarti Goyal, Sakshi Dewasthale, Meghna Gupta, Dominic Thangminlen Haokip, Sanju Barik, and et al. 2022. "Fun30 and Rtt109 Mediate Epigenetic Regulation of the DNA Damage Response Pathway in C. albicans" Journal of Fungi 8, no. 6: 559. https://doi.org/10.3390/jof8060559