
Genomic and AntiSMASH Analyses of Marine-Sponge-Derived Strain Aspergillus niger L14 Unveiling Its Vast Potential of Secondary Metabolites Biosynthesis




Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain Source
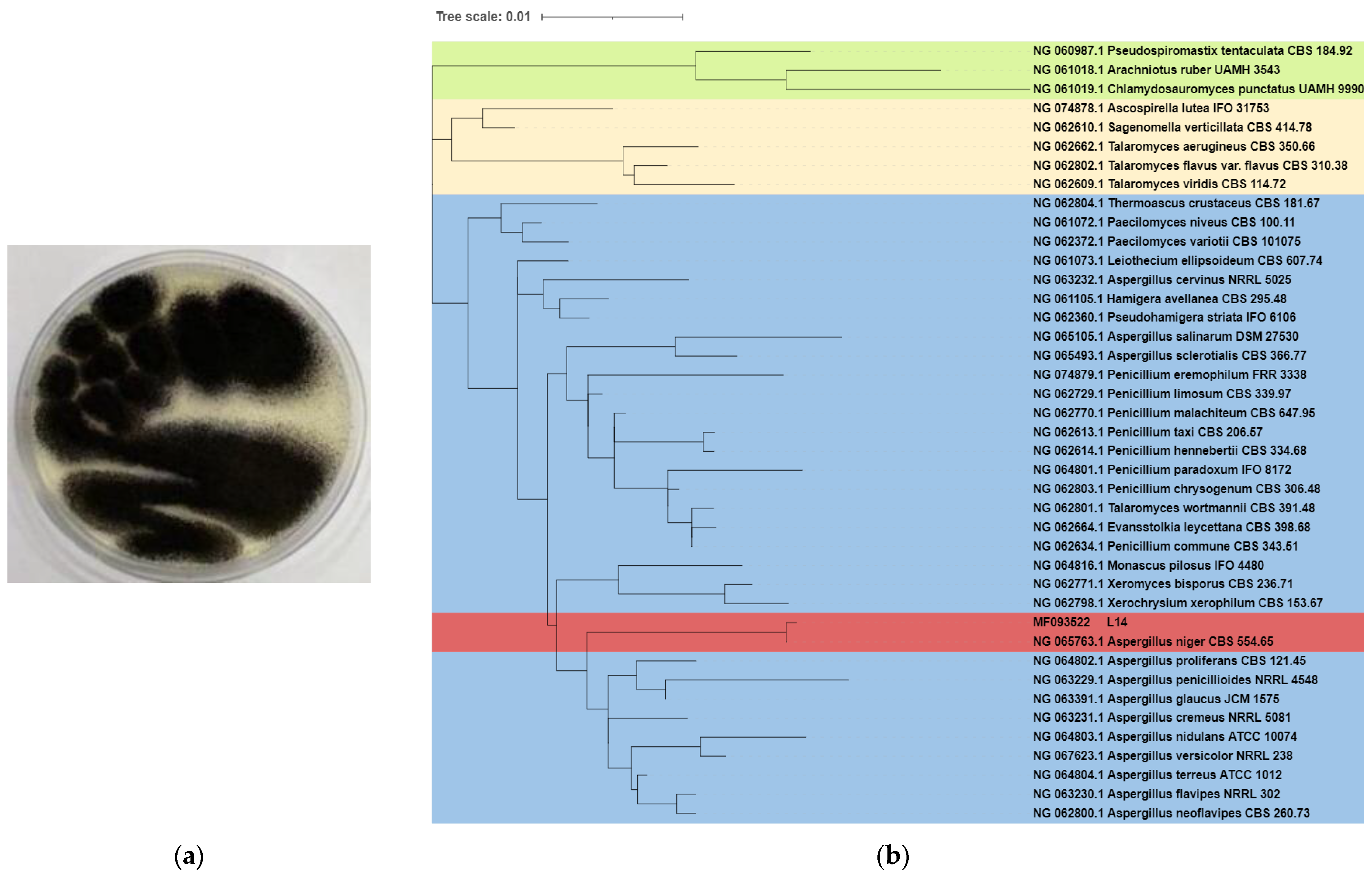
2.2. Phylogenetic Analysis
2.3. DNA Extraction and Whole Genome Sequencing
2.4. Genome Assembly
2.5. Gene Annotation
2.6. Additional Annotation
2.7. Secondary Metabolic Gene Cluster Analysis
3. Results and Discussion
3.1. Morphology and Classification of Strain L14
3.2. Genomic Assembly and Analysis
3.3. Functional Gene Annotation
3.4. Additional Annotation
3.5. AntiSMASH Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, K.; Ding, P. New bioactive metabolites from the marine-derived fungi Aspergillus. Mini-Rev. Med. Chem. 2018, 18, 1072–1094. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Li, H.; Zhang, P.; Bao, B.Q.; Lee, K.; Jung, J. Marine-derived Aspergillus species as a source of bioactive secondary metabolites. Mar. Biotechnol. 2013, 15, 499–519. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, K.; Arai, M.; Setiawan, A.; Kobayashi, M. Anti-dormant mycobacterial activity of viomellein and xanthomegnin, naphthoquinone dimers produced by marine-derived Aspergillus sp. Nat. Prod. Commun. 2017, 12, 579–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.; Han, X.; Liu, J.; Zhu, Q.; Zhang, Y.; Wang, D.; Zhang, C.; Zhao, D. Research progress on secondary metabolites with anti-phytopathogenic activities of marine-derived Aspergillus sp.and Trichoderma sp. J. Agric. Sci. 2020, 36, 1332–1341. [Google Scholar]
- Machida, M. Genome sequencing of Aspergillus oryzae. Tanpakushitsu Kakusan Koso 2006, 51, 452–456. [Google Scholar]
- Medema, M.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.; Weber, T.; Takano, E.; Breitling, R. AntiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Blin, K.; Medema, M.; Kottmann, R.; Lee, S.; Weber, T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2017, 45, D555–D559. [Google Scholar] [CrossRef] [Green Version]
- Alekseev, K.V.; Dubina, M.V.; Komov, V.P. Metabolic characteristics of citric acid synthesis by the fungus Aspergillus niger. Appl. Biochem. Microbiol. 2015, 51, 857–865. [Google Scholar] [CrossRef]
- Zhao, M.L.; Lu, X.Y.; Zong, H.; Li, J.Y.; Zhuge, B. Itaconic acid production in microorganisms. Biotechnol. Lett. 2018, 40, 455–464. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Fukuda, T.; Hagimori, K.; Tomoda, H.; Omura, S. Tensyuic acids, new antibiotics produced by Aspergillus niger FKI-2342. Chem. Pharm. Bull. 2007, 55, 1338–1341. [Google Scholar] [CrossRef] [Green Version]
- Kato, M. An overview of the CCAAT-box binding factor in filamentous fungi: Assembly, nuclear translocation, and transcriptional enhancement. Biosci. Biotechnol. Biochem. 2005, 69, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Dong, M.; Lai, T.; Zhang, H. Antimicrobial evaluation of the crude extract of symbiotic fungi from marine sponge Reniera japonica. Bangladesh J. Pharmacol. 2018, 13, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Leggett, R.; Clark, M. A world of opportunities with nanopore sequencing. J. Exp. Bot. 2017, 68, 5419–5429. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.; Judd, L.; Gorrie, C.; Holt, K. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Mu, O. Soap-HT-BLAST: High throughput BLAST based on web services. Bioinformatics 2003, 19, 1863–1864. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.D.; Mi, H.Y.; Lewis, S. Ontology annotation: Mapping genomic regions to biological function. Curr. Opin. Chem. Biol. 2007, 11, 4–11. [Google Scholar] [CrossRef]
- Li, X.; Liu, Z.; Li, J.; Fang, H. Recent progress and application of KEGG database in the research of bioinformatics. Pharm. Biotechnol. 2012, 19, 535–539. [Google Scholar]
- Oka, T.; Ungar, D.; Hughson, F.; Krieger, M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol. Biol. Cell 2004, 15, 2423–2435. [Google Scholar] [CrossRef]
- Wang, S.; Chen, G.; Zhang, H.; Wang, L. Carbohydrate-active enzyme (CAZy) database and its new prospect. Chin. J. Bioprocess Eng. 2014, 12, 102–108. [Google Scholar]
- Blin, K.; Wolf, T.; Chevrette, M.G.; Lu, X.W.; Schwalen, C.J.; Kautsar, S.A.; Duran, H.G.S.; Santos, E.; Kim, H.U.; Nave, M.; et al. AntiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017, 45, W36–W41. [Google Scholar] [CrossRef]
- Robbins, T.; Liu, Y.C.; Cane, D.E.; Khosla, C. Structure and mechanism of assembly line polyketide synthases. Curr. Opin. Struct. Biol. 2016, 41, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Pan, H.; Tang, G. New insights into bacterial type II polyketide biosynthesis. F1000Research 2017, 6, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, J.; Schoppet, M.; Hansen, M.; Cryle, M. Diversity of nature’s assembly lines—Recent discoveries in non-ribosomal peptide synthesis. Mol. Biosyst. 2017, 13, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genom. 2013, 14, 15. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chooi, Y.; Sheng, Y.; Valentine, J.; Tang, Y. Comparative characterization of fungal anthracenone and naphthacenedione biosynthetic pathways reveals an alpha-hydroxylation-dependent claisen-like cyclization catalyzed by a dimanganese thioesterase. J. Am. Chem. Soc. 2011, 133, 15773–15785. [Google Scholar] [CrossRef] [Green Version]
- Holm, D.; Petersen, L.; Klitgaard, A.; Knudsen, P.; Jarczynska, Z.; Nielsen, K.; Gotfredsen, C.; Larsen, T.; Mortensen, U. Molecular and chemical characterization of the biosynthesis of the 6-MSA-drived meroterpenoid yanuthone D in Aspergillus niger. Chem. Biol. 2014, 21, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Kihara, J.; Moriwaki, A.; Tanaka, N.; Tanaka, C.; Ueno, M.; Arase, S. Characterization of the BMR1 gene encoding a transcription factor for melanin biosynthesis genes in the phytopathogenic fungus Bipolaris oryzae. FEMS Microbiol. Lett. 2008, 281, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Kihara, J.; Moriwaki, A.; Ueno, M.; Tokunaga, T.; Arase, S.; Honda, Y. Cloning, functional analysis and expression of a scytalone dehydratase gene (SCD1) involved in melanin biosynthesis of the phytopathogenic fungus Bipolaris oryzae. Curr. Genet. 2004, 45, 197–204. [Google Scholar] [CrossRef]
- Kihara, J.; Moriwaki, A.; Ito, M.; Arase, S.; Honda, Y. Expression of THR1, a 1,3,8-trihydroxynaphthalene reductase gene involved in melanin biosynthesis in the phytopathogenic fungus Bipolaris oryzae, is enhanced by near-ultraviolet radiation. Pigm. Cell. Res. 2004, 17, 15–23. [Google Scholar] [CrossRef]
- Moriwaki, A.; Kihara, J.; Kobayashi, T.; Tokunaga, T.; Arase, S.; Honda, Y. Insertional mutagenesis and characterization of a polyketide synthase gene (PKS1) required for melanin biosynthesis in Bipolaris oryzae. FEMS Microbiol. Lett. 2004, 238, 1–8. [Google Scholar]
- Zabala, A.; Xu, W.; Chooi, Y.; Tang, Y. Characterization of a silent azaphilone gene cluster from Aspergillus niger ATCC 1015 reveals a hydroxylation-mediated pyran-ring formation. Chem. Biol. 2012, 19, 1049–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, M.; Nielsen, J.B.; Klitgaard, A.; Petersen, L.; Zachariasen, M.; Hansen, T.J.; Blicher, L.H.; Gotfredsen, C.; Larsen, T.; Nielsen, K.; et al. Accurate prediction of secondary metabolite gene clusters in filamentous fungi. Proc. Natl. Acad. Sci. USA 2013, 110, E99–E107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slightom, J.; Metzger, B.; Luu, H.; Elhammer, A. Cloning and molecular characterization of the gene encoding the aureobasidin A biosynthesis complex in Aureobasidium pullulans BP-1938. Gene 2009, 431, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Yue, Q.; Chen, L.; Li, Y.; Bills, G.; Zhang, X.; Xiang, M.; Li, S.J.; Che, Y.; Wang, C.; Niu, X.; et al. Functional operons in secondary metabolic gene clusters in glarea lozoyensis (fungi, mscomycota, leotiomycetes). mBio 2015, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Value |
|---|---|
| Total length (bp) | 36,104,262 |
| Contigs | 69 |
| GC content (%) | 49.3 |
| N50 (bp) | 4,203,503 |
| N90 (bp) | 2,686,881 |
| Max-length (bp) | 9,886,207 |
| Type | Repeat/Copy Number | Repeat Size/Total Length (bp) | Average Length (bp) | In Genome% |
|---|---|---|---|---|
| DNA | 82 | 33,600 | 409 | 0.0931 |
| LINE | 89 | 52,257 | 587 | 0.1447 |
| LTR | 166 | 79,092 | 476 | 0.2191 |
| RC | 5 | 2595 | 519 | 0.0072 |
| miRNA | 0 | 0 | 0 | 0 |
| rRNA | 80 | 70,794 | 884 | 0.1961 |
| snRNA | 34 | 4130 | 121 | 0.0114 |
| tRNA | 296 | 560 | 1 | 0.0016 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Xu, S.; Tang, Y.; Wang, H.; Bai, X.; Zhang, H. Genomic and AntiSMASH Analyses of Marine-Sponge-Derived Strain Aspergillus niger L14 Unveiling Its Vast Potential of Secondary Metabolites Biosynthesis. J. Fungi 2022, 8, 591. https://doi.org/10.3390/jof8060591
Wang P, Xu S, Tang Y, Wang H, Bai X, Zhang H. Genomic and AntiSMASH Analyses of Marine-Sponge-Derived Strain Aspergillus niger L14 Unveiling Its Vast Potential of Secondary Metabolites Biosynthesis. Journal of Fungi. 2022; 8(6):591. https://doi.org/10.3390/jof8060591
Chicago/Turabian StyleWang, Ping, Shuang Xu, Yuqi Tang, Hong Wang, Xuelian Bai, and Huawei Zhang. 2022. "Genomic and AntiSMASH Analyses of Marine-Sponge-Derived Strain Aspergillus niger L14 Unveiling Its Vast Potential of Secondary Metabolites Biosynthesis" Journal of Fungi 8, no. 6: 591. https://doi.org/10.3390/jof8060591
APA StyleWang, P., Xu, S., Tang, Y., Wang, H., Bai, X., & Zhang, H. (2022). Genomic and AntiSMASH Analyses of Marine-Sponge-Derived Strain Aspergillus niger L14 Unveiling Its Vast Potential of Secondary Metabolites Biosynthesis. Journal of Fungi, 8(6), 591. https://doi.org/10.3390/jof8060591