
Development of the CRISPR-Cas9 System for the Marine-Derived Fungi Spiromastix sp. SCSIO F190 and Aspergillus sp. SCSIO SX7S7

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Culture Conditions
2.2. DNA Manipulation
2.3. Maker Gene Selection and Fungal Sensitivity Test
2.4. Construction of Cas9 and gRNA Expression Vectors
2.5. Optimization of Protoplast Preparation
2.6. Transformation and Regeneration of Protoplasts
2.7. Transformant Validation via MSBSP-PCR and Sequencing
2.8. Secondary Metabolite Analysis
3. Results
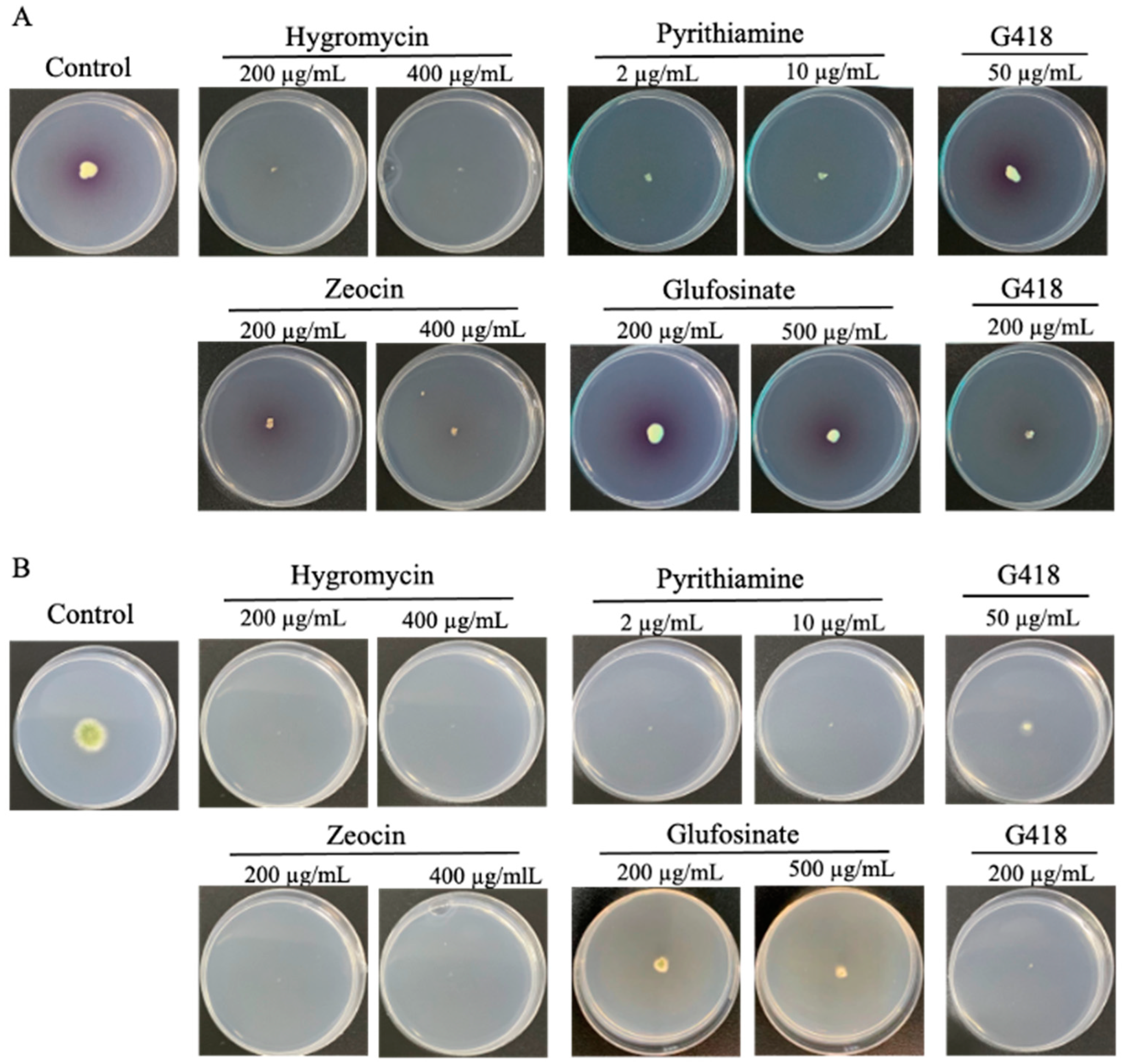
3.1. Antibiotic Sensitivity Test and Resistance Maker Gene Selection
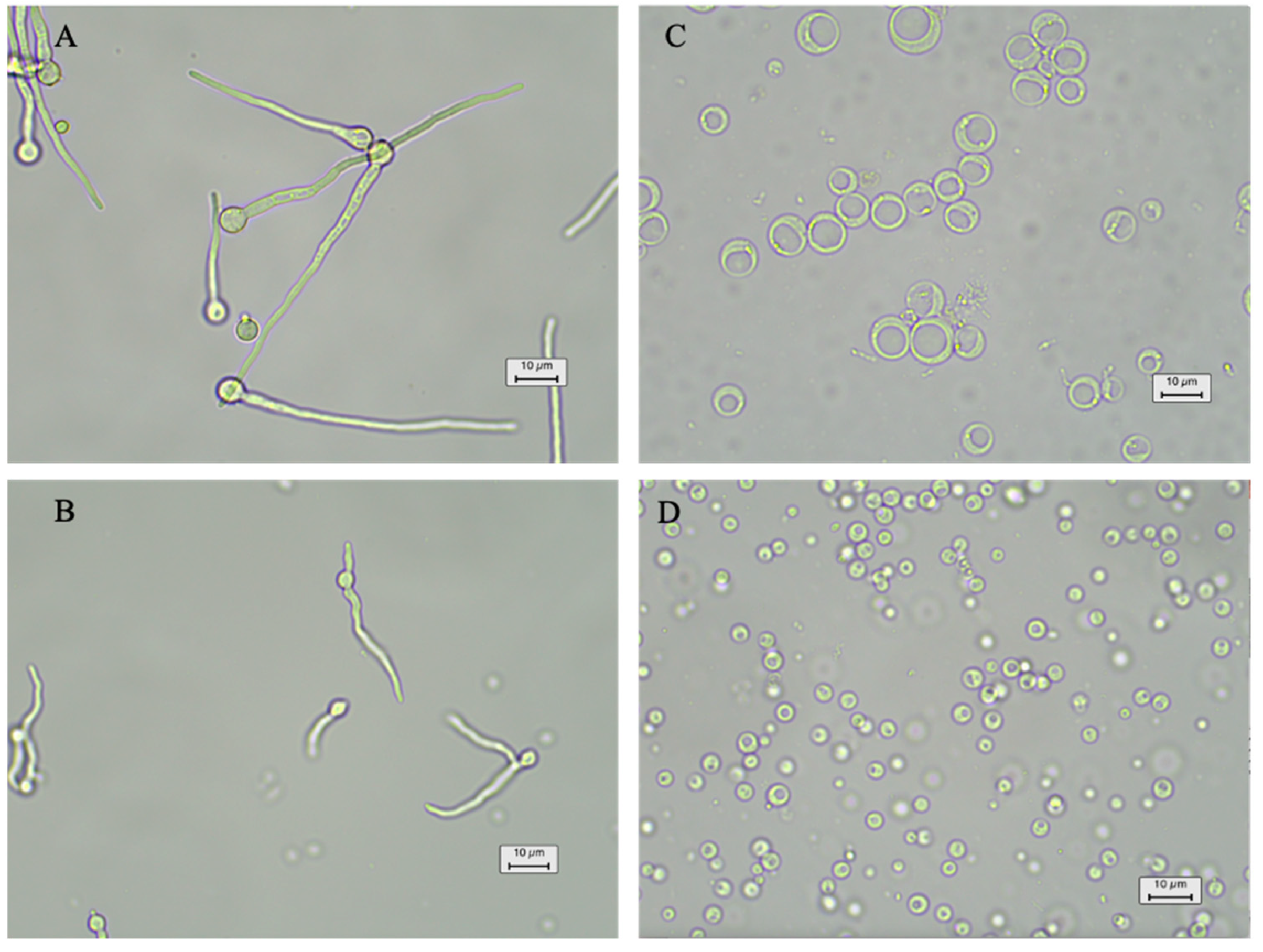
3.2. Establishment of Protoplast Preparation and Transformation
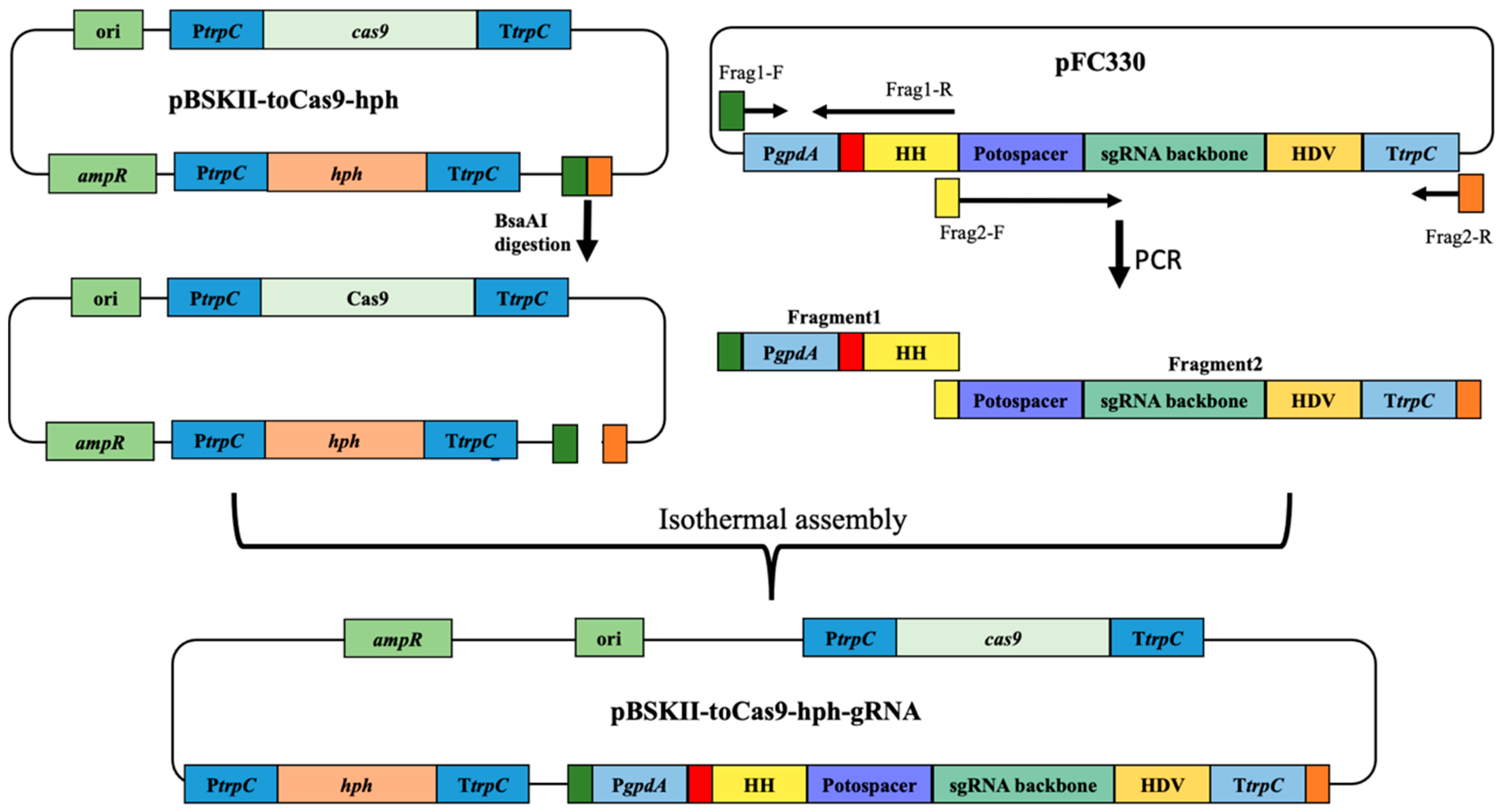
3.3. CRISPR-Cas9 Plasmid Construction for Gene Inactivation
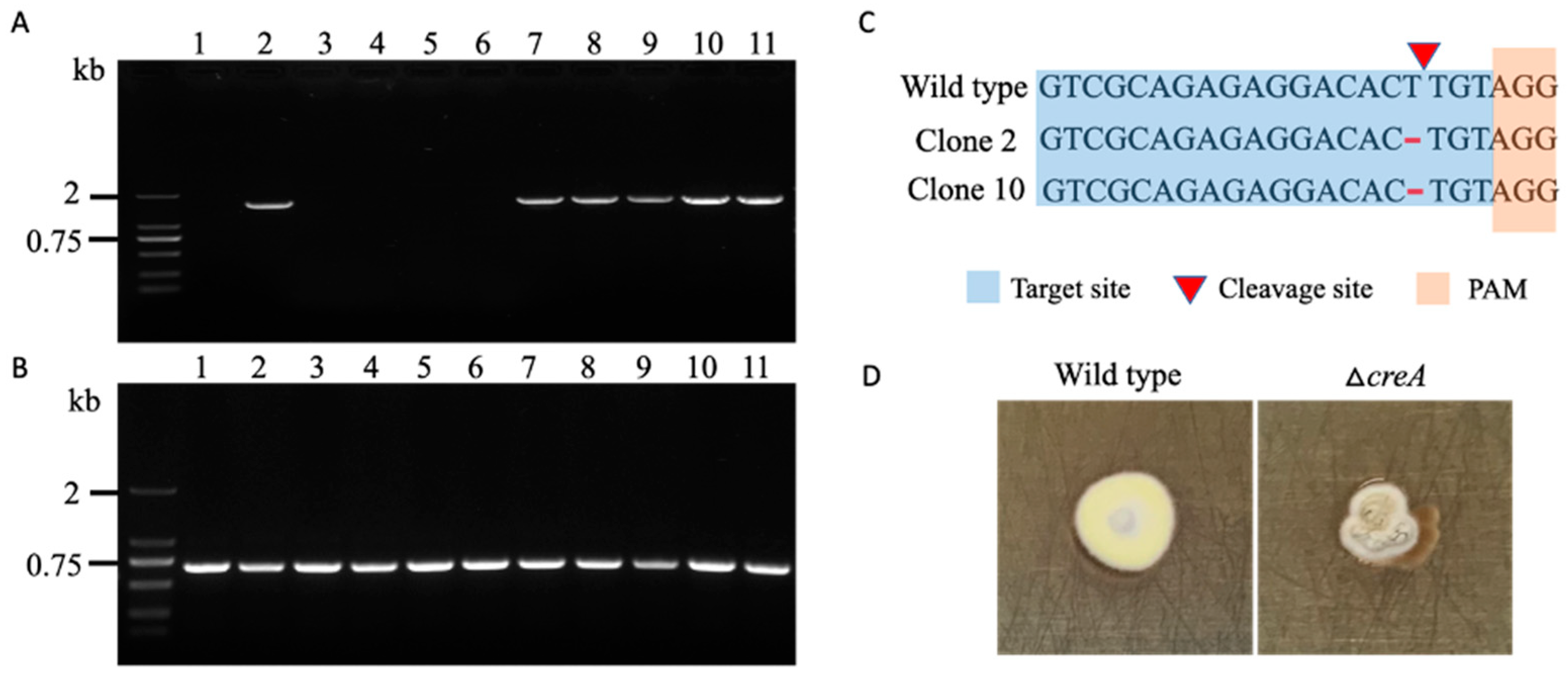
3.4. Target Gene Deletion in Spiromastix sp. SCSIO F190 and Aspergillus sp. SCSIO SX7S7
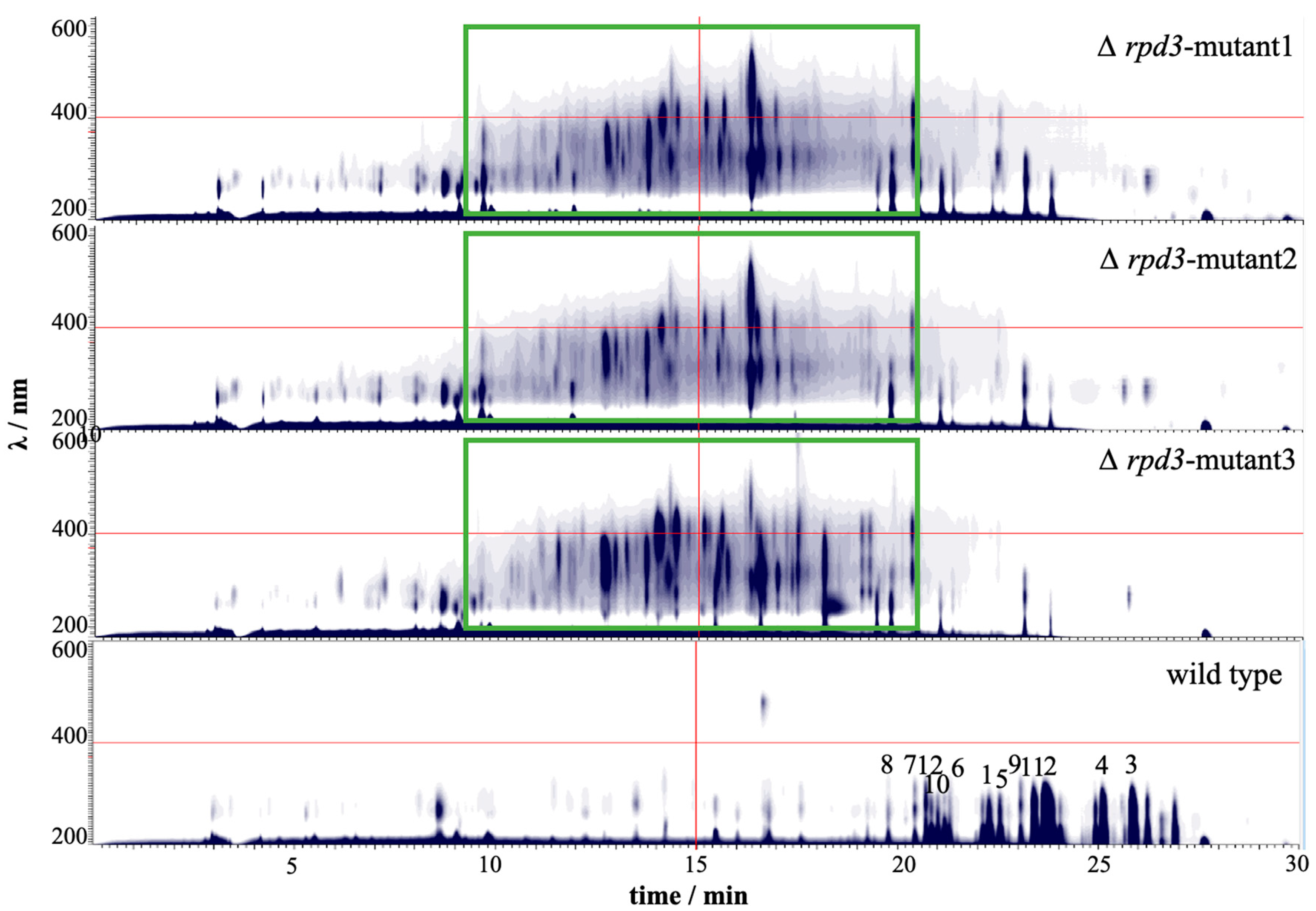
3.5. CRISPR-Cas9 Can Efficiently Mutate Histone Deacetylase Gene for Novel Natural Products Activation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hasan, S.; Ansari, M.I.; Ahmad, A.; Mishra, M. Major bioactive metabolites from marine fungi: A Review. Bioinformation 2015, 11, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Shabana, S.; Lakshmi, K.R.; Satya, A.K. An Updated Review of Secondary Metabolites from Marine Fungi. Mini Rev. Med. Chem. 2021, 21, 602–642. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.G.; Abraham, E.P. Cephalosporin C, a new antibiotic containing sulphur and D-alpha-aminoadipic acid. Nature 1955, 175, 548. [Google Scholar] [CrossRef]
- Gomes, N.G.M.; Madureira-Carvalho, A.; Dias-da-Silva, D.; Valentao, P.; Andrade, P.B. Biosynthetic versatility of marine-derived fungi on the delivery of novel antibacterial agents against priority pathogens. Biomed. Pharmacother. 2021, 140, 111756. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.K.; Prakash, V.; Ranjan, N. Marine Fungi: A Source of Potential Anticancer Compounds. Front. Microbiol. 2017, 8, 2536. [Google Scholar] [CrossRef]
- Mao, X.M.; Xu, W.; Li, D.; Yin, W.B.; Chooi, Y.H.; Li, Y.Q.; Tang, Y.; Hu, Y. Epigenetic genome mining of an endophytic fungus leads to the pleiotropic biosynthesis of natural products. Angew. Chem. Int. Ed. Engl. 2015, 54, 7592–7596. [Google Scholar] [CrossRef] [Green Version]
- Macheleidt, J.; Mattern, D.J.; Fischer, J.; Netzker, T.; Weber, J.; Schroeckh, V.; Valiante, V.; Brakhage, A.A. Regulation and Role of Fungal Secondary Metabolites. Annu. Rev. Genet. 2016, 50, 371–392. [Google Scholar] [CrossRef]
- Jiang, C.; Lv, G.; Tu, Y.; Cheng, X.; Duan, Y.; Zeng, B.; He, B. Applications of CRISPR/Cas9 in the Synthesis of Secondary Metabolites in Filamentous Fungi. Front. Microbiol. 2021, 12, 638096. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Liu, R.; Chen, L.; Jiang, Y.; Zhou, Z.; Zou, G. Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discov 2015, 1, 15007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arazoe, T.; Miyoshi, K.; Yamato, T.; Ogawa, T.; Ohsato, S.; Arie, T.; Kuwata, S. Tailor-made CRISPR/Cas system for highly efficient targeted gene replacement in the rice blast fungus. Biotechnol. Bioeng. 2015, 112, 2543–2549. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T.; Tanaka, Y.; Okabe, T.; Nakamura, H.; Fujii, W.; Kitamoto, K.; Maruyama, J. Development of a genome editing technique using the CRISPR/Cas9 system in the industrial filamentous fungus Aspergillus oryzae. Biotechnol. Lett. 2016, 38, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Kuivanen, J.; Wang, Y.J.; Richard, P. Engineering Aspergillus niger for galactaric acid production: Elimination of galactaric acid catabolism by using RNA sequencing and CRISPR/Cas9. Microb. Cell Fact. 2016, 15, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, K.K.; Chen, S.; Loros, J.J.; Dunlap, J.C. Development of the CRISPR/Cas9 System for Targeted Gene Disruption in Aspergillus fumigatus. Eukaryot. Cell 2015, 14, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Nodvig, C.S.; Nielsen, J.B.; Kogle, M.E.; Mortensen, U.H. A CRISPR-Cas9 System for Genetic Engineering of Filamentous Fungi. PLoS ONE 2015, 10, e0133085. [Google Scholar]
- Nielsen, M.L.; Isbrandt, T.; Rasmussen, K.B.; Thrane, U.; Hoof, J.B.; Larsen, T.O.; Mortensen, U.H. Genes Linked to Production of Secondary Metabolites in Talaromyces atroroseus Revealed Using CRISPR-Cas9. PLoS ONE 2017, 12, e0169712. [Google Scholar] [CrossRef] [Green Version]
- Matsu-Ura, T.; Baek, M.; Kwon, J.; Hong, C. Efficient gene editing in Neurospora crassa with CRISPR technology. Fungal. Biol. Biotechnol. 2015, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.; Dean, N. Dramatic Improvement of CRISPR/Cas9 Editing in Candida albicans by Increased Single Guide RNA Expression. mSphere 2017, 2, e00385-16. [Google Scholar] [CrossRef] [Green Version]
- Wenderoth, M.; Pinecker, C.; Voss, B.; Fischer, R. Establishment of CRISPR/Cas9 in Alternaria alternata. Fungal. Genet. Biol. 2017, 101, 55–60. [Google Scholar] [CrossRef]
- Pohl, C.; Kiel, J.A.; Driessen, A.J.; Bovenberg, R.A.; Nygard, Y. CRISPR/Cas9 Based Genome Editing of Penicillium chrysogenum. ACS Synth. Biol. 2016, 5, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Schuster, M.; Schweizer, G.; Reissmann, S.; Kahmann, R. Genome editing in Ustilago maydis using the CRISPR-Cas system. Fungal. Genet. Biol. 2016, 89, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, J.; Duan, C.; Pan, Y.; Liu, G. Improvement of the CRISPR-Cas9 mediated gene disruption and large DNA fragment deletion based on a chimeric promoter in Acremonium chrysogenum. Fungal. Genet. Biol. 2020, 134, 103279. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.Y.; Wu, Y.J.; Xie, Q.P.; Tang, J.W.; Yu, Z.T.; Yang, S.B.; Chen, S.X. CRISPR/Cas9-Based Genome Editing in the Filamentous Fungus Glarea lozoyensis and Its Application in Manipulating gloF. ACS Synth. Biol. 2020, 9, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.M.; Lin, F.L.; Gao, H.; Zou, G.; Zhang, J.W.; Wang, G.Q.; Chen, G.D.; Zhou, Z.H.; Yao, X.S.; Hu, D. Development of a versatile and conventional technique for gene disruption in filamentous fungi based on CRISPR-Cas9 technology. Sci. Rep. 2017, 7, 9250. [Google Scholar] [CrossRef] [Green Version]
- Shao, M.; Sun, C.; Liu, X.; Wang, X.; Li, W.; Wei, X.; Li, Q.; Ju, J. Upregulation of a marine fungal biosynthetic gene cluster by an endobacterial symbiont. Commun. Biol. 2020, 3, 527. [Google Scholar] [CrossRef]
- Yang, J.F.; Zhou, L.; Zhou, Z.B.; Song, Y.X.; Ju, J.H. Anti-pathogenic depsidones and its derivatives from a coral-derived fungus Aspergillus sp. SCSIO SX7S7. Biochem. Syst. Ecol. 2022, 102, 104415. [Google Scholar] [CrossRef]
- Todd, R.B.; Davis, M.A.; Hynes, M.J. Genetic manipulation of Aspergillus nidulans: Meiotic progeny for genetic analysis and strain construction. Nat. Protoc. 2007, 2, 811–821. [Google Scholar] [CrossRef]
- Guo, J.; Li, K.; Jin, L.; Xu, R.; Miao, K.; Yang, F.; Qi, C.; Zhang, L.; Botella, J.R.; Wang, R.; et al. A simple and cost-effective method for screening of CRISPR/Cas9-induced homozygous/biallelic mutants. Plant Methods 2018, 14, 40. [Google Scholar] [CrossRef]
- Li, D.; Tang, Y.; Lin, J.; Cai, W. Methods for genetic transformation of filamentous fungi. Microb. Cell Fact. 2017, 16, 168. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Li, W.; Qi, L.; Chen, G.; Cai, L.; Yin, W.B. Establishment of a Genetic Transformation System in Guanophilic Fungus Amphichorda guana. J. Fungi 2021, 7, 138. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dong, L.; Alam, M.A.; Pardeshi, L.; Miao, Z.; Wang, F.; Tan, K.; Hynes, M.J.; Kelly, J.M.; Wong, K.H. Carbon Catabolite Repression Governs Diverse Physiological Processes and Development in Aspergillus nidulans. mBio 2022, 13, e0373421. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.P.; Hashmi, S.B.; Osmani, A.H.; Andrews, P.; Ringelberg, C.S.; Dunlap, J.C.; Osmani, S.A. Functional analysis of the Aspergillus nidulans kinome. PLoS ONE 2013, 8, e58008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makela, M.R.; Aguilar-Pontes, M.V.; van Rossen-Uffink, D.; Peng, M.; de Vries, R.P. The fungus Aspergillus niger consumes sugars in a sequential manner that is not mediated by the carbon catabolite repressor CreA. Sci. Rep. 2018, 8, 6655. [Google Scholar] [CrossRef] [Green Version]
- Jonkers, W.; Rep, M. Mutation of CRE1 in Fusarium oxysporum reverts the pathogenicity defects of the FRP1 deletion mutant. Mol. Microbiol. 2009, 74, 1100–1113. [Google Scholar] [CrossRef]
- Guzman-Chavez, F.; Salo, O.; Samol, M.; Ries, M.; Kuipers, J.; Bovenberg, R.A.L.; Vreeken, R.J.; Driessen, A.J.M. Deregulation of secondary metabolism in a histone deacetylase mutant of Penicillium chrysogenum. Microbiologyopen 2018, 7, e00598. [Google Scholar] [CrossRef] [Green Version]
- Shwab, E.K.; Bok, J.W.; Tribus, M.; Galehr, J.; Graessle, S.; Keller, N.P. Histone deacetylase activity regulates chemical diversity in Aspergillus. Eukaryot. Cell 2007, 6, 1656–1664. [Google Scholar] [CrossRef] [Green Version]
- Collemare, J.; Seidl, M.F. Chromatin-dependent regulation of secondary metabolite biosynthesis in fungi: Is the picture complete? FEMS Microbiol. Rev. 2019, 43, 591–607. [Google Scholar] [CrossRef] [Green Version]
- Amend, A.; Burgaud, G.; Cunliffe, M.; Edgcomb, V.P.; Ettinger, C.L.; Gutierrez, M.H.; Heitman, J.; Hom, E.F.Y.; Ianiri, G.; Jones, A.C.; et al. Fungi in the Marine Environment: Open Questions and Unsolved Problems. mBio 2019, 10, e01189-18. [Google Scholar] [CrossRef] [Green Version]
- Yao, G.; Chen, X.; Han, Y.; Zheng, H.; Wang, Z.; Chen, J. Development of versatile and efficient genetic tools for the marine-derived fungus Aspergillus terreus RA2905. Curr. Genet. 2022, 68, 153–164. [Google Scholar] [CrossRef]
- Ullah, M.; Xia, L.; Xie, S.; Sun, S. CRISPR/Cas9-based genome engineering: A new breakthrough in the genetic manipulation of filamentous fungi. Biotechnol. Appl. Biochem. 2020, 67, 835–851. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Zhai, Q.; Sun, L.; Huang, E.; Zhang, Y.; Zhu, Y.; Guo, Q.; Tian, Y.; Zhao, B.; Lu, H. CRISPR/Cas9 genome editing technology in filamentous fungi: Progress and perspective. Appl. Microbiol. Biotechnol. 2019, 103, 6919–6932. [Google Scholar] [CrossRef] [Green Version]
- Nissim, L.; Perli, S.D.; Fridkin, A.; Perez-Pinera, P.; Lu, T.K. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol. Cell 2014, 54, 698–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruckner, B.; Unkles, S.E.; Weltring, K.; Kinghorn, J.R. Transformation of Gibberella fujikuroi: Effect of the Aspergillus nidulans AMA1 sequence on frequency and integration. Curr. Genet. 1992, 22, 313–316. [Google Scholar] [CrossRef]
- Feng, Z.; Mao, Y.; Xu, N.; Zhang, B.; Wei, P.; Yang, D.L.; Wang, Z.; Zhang, Z.; Zheng, R.; Yang, L.; et al. Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, 4632–4637. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Roberts, H.M.; Van Eck, J.; Martin, G.B. Generation and Molecular Characterization of CRISPR/Cas9-Induced Mutations in 63 Immunity-Associated Genes in Tomato Reveals Specificity and a Range of Gene Modifications. Front. Plant Sci. 2020, 11, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, Y.; Zhang, Y.; Yu, P.L.; Pan, H.; Rollins, J.A. Introduction of Large Sequence Inserts by CRISPR-Cas9 to Create Pathogenicity Mutants in the Multinucleate Filamentous Pathogen Sclerotinia sclerotiorum. mBio 2018, 9, e00567-18. [Google Scholar] [CrossRef] [Green Version]
- Cradick, T.J.; Fine, E.J.; Antico, C.J.; Bao, G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013, 41, 9584–9592. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [Green Version]
- Brakhage, A.A. Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 2013, 11, 21–32. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Cai, C.; Yang, J.; Shi, J.; Song, Y.; Hu, D.; Ma, J.; Ju, J. Development of the CRISPR-Cas9 System for the Marine-Derived Fungi Spiromastix sp. SCSIO F190 and Aspergillus sp. SCSIO SX7S7. J. Fungi 2022, 8, 715. https://doi.org/10.3390/jof8070715
Chen Y, Cai C, Yang J, Shi J, Song Y, Hu D, Ma J, Ju J. Development of the CRISPR-Cas9 System for the Marine-Derived Fungi Spiromastix sp. SCSIO F190 and Aspergillus sp. SCSIO SX7S7. Journal of Fungi. 2022; 8(7):715. https://doi.org/10.3390/jof8070715
Chicago/Turabian StyleChen, Yingying, Cunlei Cai, Jiafan Yang, Junjie Shi, Yongxiang Song, Dan Hu, Junying Ma, and Jianhua Ju. 2022. "Development of the CRISPR-Cas9 System for the Marine-Derived Fungi Spiromastix sp. SCSIO F190 and Aspergillus sp. SCSIO SX7S7" Journal of Fungi 8, no. 7: 715. https://doi.org/10.3390/jof8070715
APA StyleChen, Y., Cai, C., Yang, J., Shi, J., Song, Y., Hu, D., Ma, J., & Ju, J. (2022). Development of the CRISPR-Cas9 System for the Marine-Derived Fungi Spiromastix sp. SCSIO F190 and Aspergillus sp. SCSIO SX7S7. Journal of Fungi, 8(7), 715. https://doi.org/10.3390/jof8070715