
Identification of Gene Modules and Hub Genes Associated with Sporisorium scitamineum Infection Using Weighted Gene Co-Expression Network Analysis

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Materials and S. scitamineum Inoculation
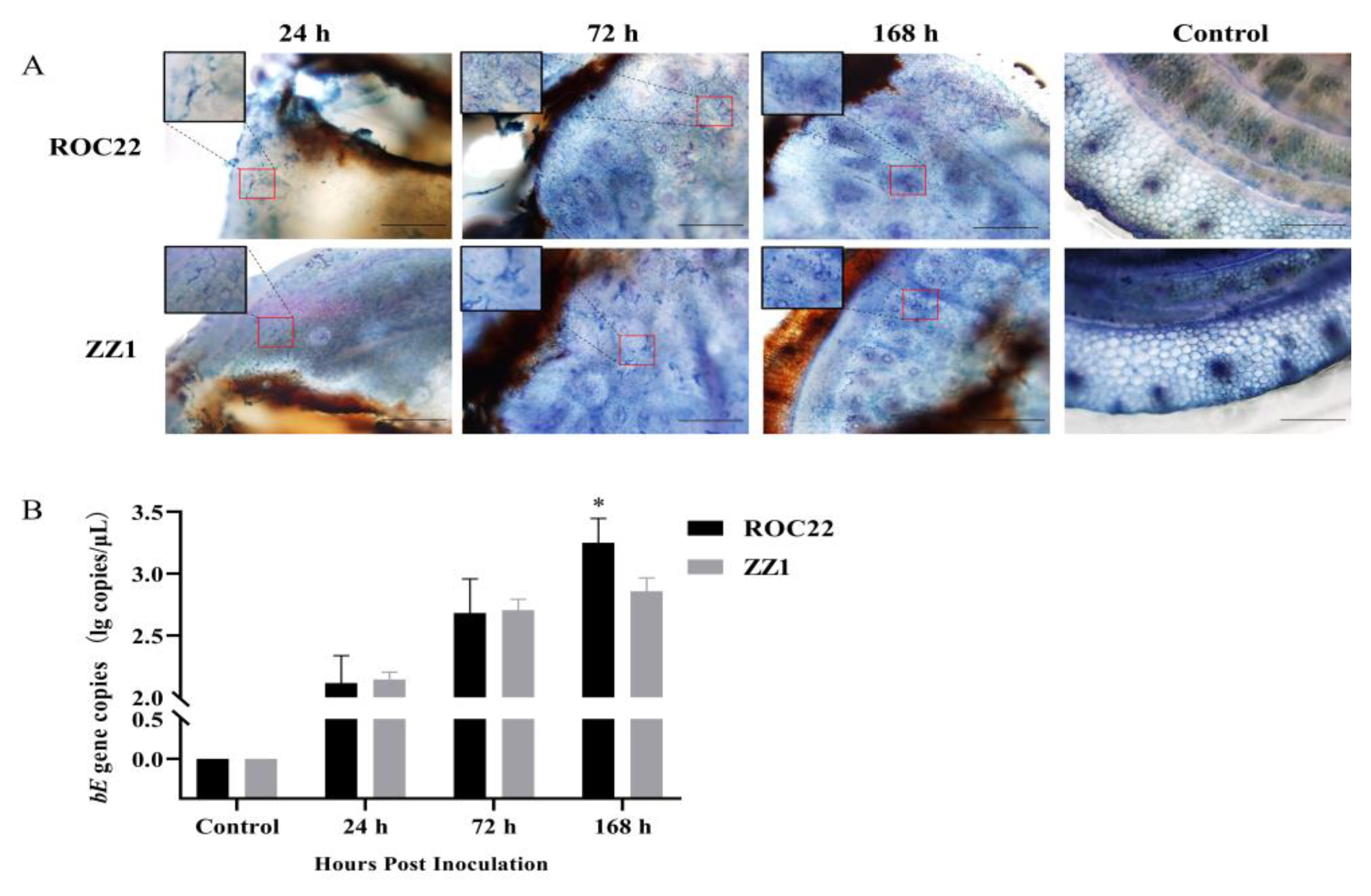
2.2. Visualization and Quantification of S. scitamineum in Plants
2.3. RNA Extraction, Library Construction, and Sequencing
2.4. Analysis of RNA-Seq Data
2.5. Effector Prediction
2.6. Reverse-Transcription Quantitative PCR (RT-qPCR)
3. Results
3.1. Colonization of S. scitamineum in Sugarcane Sprouts
3.2. Analysis of RNA-Seq Data
3.3. Identification of Modules Associated with Infection
3.4. Annotation of Genes in the Modules
3.5. Hub Genes in the Modules
3.6. Proteins in MEpurple and MEdarkturquoise Modules Related to Virulence
3.7. Candidate Effectors
4. Discussion
4.1. WGCNA Is a Powerful Tool for Screening Modules and Hub Genes Related to Fungal Infection
4.2. Endoplasmic Reticulum and Catabolism Play Essential Roles in S. scitamineum Infection
4.3. Mechanism for the Systematic Infection of Sugarcane Smut
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Croft, B.J.; Magarey, R.C.; Quinn, B.; Royal, A.; Kerkwyk, R. Sugarcane smut in Queensland. Aust. Soc. SugarCane Technol. 2008, 30, 52–62. [Google Scholar]
- Taniguti, L.M.; Schaker, P.D.; Benevenuto, J.; Peters, L.P.; Carvalho, G.; Palhares, A.; Quecine, M.C.; Nunes, F.R.; Kmit, M.C.; Wai, A.; et al. Complete Genome Sequence of Sporisorium scitamineum and Biotrophic Interaction Transcriptome with Sugarcane. PLoS ONE 2015, 10, e129318. [Google Scholar] [CrossRef]
- Vicente, C.; Legaz, M.; Sánchez-Elordi, E. Physiological Basis of Smut Infectivity in the Early Stages of Sugar Cane Colonization. J. Fungi 2021, 7, 44. [Google Scholar] [CrossRef]
- Stergiopoulos, I.; de Wit, P.J. Fungal effector proteins. Annu. Rev. Phytopathol. 2009, 47, 233–263. [Google Scholar] [CrossRef]
- De Wit, P.J.G.M.; Mehrabi, R.; Van Den Burg, H.A.; Stergiopoulos, I. Fungal effector proteins: Past, present and future. Mol. Plant Pathol. 2009, 10, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Que, Y.; Xu, L.; Wu, Q.; Liu, Y.; Ling, H.; Liu, Y.; Zhang, Y.; Guo, J.; Su, Y.; Chen, J.; et al. Genome sequencing of Sporisorium scitamineum provides insights into the pathogenic mechanisms of sugarcane smut. BMC Genom. 2014, 15, 996. [Google Scholar] [CrossRef]
- Ling, H.; Fu, X.; Huang, N.; Zhong, Z.; Su, W.; Lin, W.; Cui, H.; Que, Y. A sugarcane smut fungus effector simulates the host endogenous elicitor peptide to suppress plant immunity. New Phytol. 2022, 233, 919–933. [Google Scholar] [CrossRef]
- Teixeira-Silva, N.S.; Schaker, P.; Rody, H.; Maia, T.; Garner, C.M.; Gassmann, W.; Monteiro-Vitorello, C.B. Leaping into the Unknown World of Sporisorium scitamineum Candidate Effectors. J. Fungi 2020, 6, 339. [Google Scholar] [CrossRef]
- Maia, T.; Rody, H.; Bombardelli, R.; Souto, T.G.; Camargo, L.; Monteiro-Vitorello, C.B. A bacterial type three secretion-based delivery system for functional characterization of Sporisorium scitamineum plant immune suppressing effector proteins. Phytopathology 2022, 112, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Peng, J.; Wang, P.; Fang, H.; Zheng, J.; Zhong, C.; Yang, Y.; Yu, W. Weighted Gene Co-Expression Analysis Network-Based Analysis on the Candidate Pathways and Hub Genes in Eggplant Bacterial Wilt-Resistance: A Plant Research Study. Int. J. Mol. Sci. 2021, 22, 3279. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, R.; Li, D.; Wang, F. Transcriptomic and Coexpression Network Analyses Revealed Pine Chalcone Synthase Genes Associated with Pine Wood Nematode Infection. Int. J. Mol. Sci. 2021, 22, 11195. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Zhang, J.; Zhang, L.; Hou, Y.; Pu, S.; Chu, A.; Bai, M.; Zhang, Z. Weighted Gene Co-Expression Network Analysis Identifies Critical Genes in the Development of Heart Failure After Acute Myocardial Infarction. Front. Genet. 2019, 10, 1214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, P.; Lan, X.; Li, R.; Huang, Y.; Chen, B. Identification of smut resistance in sugarcane Zhongzhe cultivars. Sugar Crops China 2019, 41, 37–40. [Google Scholar] [CrossRef]
- Lu, S.; Guo, F.; Wang, Z.; Shen, X.; Deng, Y.; Meng, J.; Jiang, Z.; Chen, B. Genetic Dissection of T-DNA Insertional Mutants Reveals Uncoupling of Dikaryotic Filamentation and Virulence in Sugarcane Smut Fungus. Phytopathology 2021, 111, 2303–2308. [Google Scholar] [CrossRef]
- Que, Y.; Su, Y.; Guo, J.; Wu, Q.; Xu, L. A global view of transcriptome dynamics during Sporisorium scitamineum challenge in sugarcane by RNA-Seq. PLoS ONE 2014, 9, e106476. [Google Scholar] [CrossRef]
- Su, Y.; Wang, S.; Guo, J.; Xue, B.; Xu, L.; Que, Y. A TaqMan real-time PCR assay for detection and quantification of Sporisorium scitamineum in sugarcane. Sci. World J. 2013, 2013, 942682. [Google Scholar] [CrossRef]
- Su, Y.; Wang, Z.; Xu, L.; Peng, Q.; Liu, F.; Li, Z.; Que, Y. Early Selection for Smut Resistance in Sugarcane Using Pathogen Proliferation and Changes in Physiological and Biochemical Indices. Front. Plant Sci. 2016, 7, 1133. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Ma, H.; He, Z.; Chen, J.; Zhang, X.; Song, P. Identifying of biomarkers associated with gastric cancer based on 11 topological analysis methods of CytoHubba. Sci. Rep. 2021, 11, 1331. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tian, L.; Zhang, D.D.; Song, J.; Song, S.S.; Yin, C.M.; Zhou, L.; Liu, Y.; Wang, B.L.; Kong, Z.Q.; et al. Functional analyses of small secreted cysteine-rich proteins identified candidate effectors in Verticillium dahliae. Mol. Plant Pathol. 2020, 21, 667–685. [Google Scholar] [CrossRef]
- Liu, Z.; Lan, X.; Li, X.; Zhao, H.; Gan, J.; Li, R.; Chen, B. A Plant-Derived Alkanol Induces Teliospore Germination in Sporisorium scitamineum. J. Fungi 2022, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bhuiyan, S.A.; Magarey, R.C.; McNeil, M.D.; Aitken, K.S. Sugarcane Smut, Caused by Sporisorium scitamineum, a Major Disease of Sugarcane: A Contemporary Review. Phytopathology 2021, 111, 1905–1917. [Google Scholar] [CrossRef]
- Liu, G.L.; Wang, D.S.; Wang, L.F.; Zhao, S.F.; Chi, Z.M. Mig1 is involved in mycelial formation and expression of the genes encoding extracellular enzymes in Saccharomycopsis fibuligera A11. Fungal Genet. Biol. 2011, 48, 904–913. [Google Scholar] [CrossRef]
- Sprockett, D.D.; Piontkivska, H.; Blackwood, C.B. Evolutionary analysis of glycosyl hydrolase family 28 (GH28) suggests lineage-specific expansions in necrotrophic fungal pathogens. Gene 2011, 479, 29–36. [Google Scholar] [CrossRef]
- López-Mondéjar, R.; Catalano, V.; Kubicek, C.P.; Seidl, V. The β-N-acetylglucosaminidases NAG1 and NAG2 are essential for growth of Trichoderma atroviride on chitin. FEBS J. 2009, 276, 5137–5148. [Google Scholar] [CrossRef] [PubMed]
- Doehlemann, G.; Reissmann, S.; Assmann, D.; Fleckenstein, M.; Kahmann, R. Two linked genes encoding a secreted effector and a membrane protein are essential for Ustilago maydis-induced tumour formation. Mol. Microbiol. 2011, 81, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Bolger, T.A.; Folkmann, A.W.; Tran, E.J.; Wente, S.R. The mRNA Export Factor Gle1 and Inositol Hexakisphosphate Regulate Distinct Stages of Translation. Cell 2008, 134, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Harlan, J.E.; Hajduk, P.J.; Yoon, H.S.; Fesik, S.W. Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature 1994, 371, 168–170. [Google Scholar] [CrossRef]
- Askew, D.S. Endoplasmic reticulum stress and fungal pathogenesis converge. Virulence 2014, 5, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.; Sasse, C.; Schedler, A.; Hasenberg, M.; Gunzer, M.; Krappmann, S. Shaping the fungal adaptome--stress responses of Aspergillus fumigatus. Int. J. Med. Microbiol. 2011, 301, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.A.; Hollien, J. The unfolded protein response in secretory cell function. Annu. Rev. Genet. 2012, 46, 165–183. [Google Scholar] [CrossRef]
- Kitamura, M. The unfolded protein response triggered by environmental factors. Semin. Immunopathol. 2013, 35, 259–275. [Google Scholar] [CrossRef]
- Nagendran, S.; Hallen-Adams, H.E.; Paper, J.M.; Aslam, N.; Walton, J.D. Reduced genomic potential for secreted plant cell-wall-degrading enzymes in the ectomycorrhizal fungus Amanita bisporigera, based on the secretome of Trichoderma reesei. Fungal Genet. Biol. 2009, 46, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Liou, R. Two genes encoding GH10 xylanases are essential for the virulence of the oomycete plant pathogen Phytophthora parasitica. Curr. Genet. 2018, 64, 931–943. [Google Scholar] [CrossRef]
- Pukkila-Worley, R.; Gerrald, Q.D.; Kraus, P.R.; Boily, M.J.; Davis, M.J.; Giles, S.S.; Cox, G.M.; Heitman, J.; Alspaugh, J.A. Transcriptional network of multiple capsule and melanin genes governed by the Cryptococcus neoformans cyclic AMP cascade. Eukaryot. Cell 2005, 4, 190–201. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-base: The pathogen-host interactions database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Y.; Shen, X.; Guo, F.; Zhou, C.; Li, R.; Chen, B. SsPEP1, an Effector with Essential Cellular Functions in Sugarcane Smut Fungus. J. Fungi 2021, 7, 954. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.; Shen, C.; Fu, Y.; Xie, J.; Jiang, D.; Li, G.; Cheng, J. A Small Secreted Virulence-Related Protein Is Essential for the Necrotrophic Interactions of Sclerotinia sclerotiorum with Its Host Plants. PLoS Pathog. 2016, 12, e1005435. [Google Scholar] [CrossRef]
- Djamei, A.; Schipper, K.; Rabe, F.; Ghosh, A.; Vincon, V.; Kahnt, J.; Osorio, S.; Tohge, T.; Fernie, A.R.; Feussner, I.; et al. Metabolic priming by a secreted fungal effector. Nature 2011, 478, 395–398. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Hof, A.; Zechmann, B.; Schwammbach, D.; Hückelhoven, R.; Doehlemann, G. Alternative Cell Death Mechanisms Determine Epidermal Resistance in Incompatible Barley-Ustilago Interactions. Mol. Plant Microbe 2013, 27, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ni, H.; Du, X.; Wang, S.; Ma, X.; Nürnberger, T.; Guo, H.; Hua, C. The Verticillium-specific protein VdSCP7 localizes to the plant nucleus and modulates immunity to fungal infections. New Phytol. 2017, 215, 368–381. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, R.; van Esse, H.P.; Maruthachalam, K.; Bolton, M.D.; Santhanam, P.; Saber, M.K.; Zhang, Z.; Usami, T.; Lievens, B.; Subbarao, K.V.; et al. Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 5110–5115. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Upregulated | Downregulated | All |
|---|---|---|---|
| TROC22_24 h vs. 0 h | 763 | 34 | 797 |
| TROC22_72 h vs. 0 h | 2887 | 32 | 2919 |
| TROC22_168 h vs. 0 h | 705 | 285 | 990 |
| TZZ1_24 h vs. 0 h | 896 | 330 | 1226 |
| TZZ1_72 h vs. 0 h | 2240 | 181 | 2421 |
| TZZ1_168 h vs. 0 h | 1225 | 261 | 1486 |
| Gene ID | Gene Module | Description | Degree | CDS Length |
|---|---|---|---|---|
| SPSC_04270 | MEpurple | Mig1-Mig1 protein | 42 | 203 |
| SPSC_03768 | MEpurple | Uncharacterized protein | 36 | 571 |
| SPSC_06609 | MEpurple | Uncharacterized protein | 35 | 133 |
| SPSC_00576 | MEpurple | Uncharacterized protein | 34 | 403 |
| SPSC_06362 | MEpurple | Uncharacterized protein | 33 | 1745 |
| SPSC_05923 | MEpurple | Glycosyl hydrolase | 33 | 737 |
| SPSC_01958 | MEpurple | Beta-N-acetylglucosaminidase | 33 | 703 |
| SPSC_04676 | MEpurple | Uncharacterized protein | 33 | 180 |
| SPSC_02155 | MEpurple | Secreted chorismate mutase | 33 | 303 |
| SPSC_04321 | MEpurple | Collagen | 33 | 584 |
| SPSC_00606 | MEdarkturquoise | Uncharacterized protein | 17 | 125 |
| SPSC_02450 | MEdarkturquoise | Uncharacterized protein | 17 | 75 |
| SPSC_05681 | MEdarkturquoise | Uncharacterized protein | 16 | 911 |
| SPSC_01622 | MEdarkturquoise | Uncharacterized protein | 16 | 300 |
| SPSC_00571 | MEdarkturquoise | Transcription and mRNA export factor | 14 | 113 |
| SPSC_01364 | MEdarkturquoise | Uncharacterized protein | 14 | 118 |
| SPSC_00940 | MEdarkturquoise | Pleckstrin homology domain protein | 14 | 1428 |
| SPSC_05327 | MEdarkturquoise | Uncharacterized protein | 13 | 167 |
| SPSC_03542 | MEdarkturquoise | Uncharacterized protein | 13 | 370 |
| SPSC_03041 | MEdarkturquoise | Uncharacterized protein | 12 | 342 |
| Gene ID | Uniprot Annotation | Length (AA) | Cysteine Percentage (%) | Signal Peptide Likelihood | Signal Peptide Position | Location | Transmembrane Domain Numbers |
|---|---|---|---|---|---|---|---|
| SPSC_06609 | Uncharacterized protein | 133 | 6.0150 | 0.9998 | pos: 26–27 | Extracellular (Secreted) | 0 |
| SPSC_02061 | Uncharacterized protein | 157 | 5.0955 | 0.9997 | pos: 23–24 | Extracellular (Secreted) | 0 |
| SPSC_01514 | Related to Endoglucanase 1 | 311 | 4.8232 | 0.9997 | pos: 29–30 | Extracellular (Secreted) | 0 |
| SPSC_01234 | Uncharacterized protein | 136 | 4.4118 | 0.9998 | pos: 24–25 | Extracellular (Secreted) | 0 |
| SPSC_01238 | Uncharacterized protein | 144 | 4.1667 | 0.9998 | pos: 21–22 | Extracellular (Secreted) | 0 |
| SPSC_02060 | Uncharacterized protein | 194 | 4.1237 | 0.9998 | pos: 25–26 | Extracellular (Secreted) | 0 |
| SPSC_06022 | Probable Endoglucanase 1 | 380 | 3.9474 | 0.9998 | pos: 26–27 | Extracellular (Secreted) | 0 |
| SPSC_04237 | Uncharacterized protein | 130 | 3.8462 | 0.9997 | pos: 21–22 | Extracellular (Secreted) | 0 |
| SPSC_04668 | Uncharacterized protein | 132 | 3.7879 | 0.9997 | pos: 24–25 | Extracellular (Secreted) | 0 |
| SPSC_05055 | Uncharacterized protein | 80 | 3.7500 | 0.9970 | pos: 32–33 | Extracellular (Secreted) | 0 |
| SPSC_04238 | Uncharacterized protein | 136 | 3.6765 | 0.9998 | pos: 22–23 | Extracellular (Secreted) | 0 |
| SPSC_04236 | Uncharacterized protein | 229 | 3.4934 | 0.9998 | pos: 21–22 | Extracellular (Secreted) | 0 |
| SPSC_02851 | Uncharacterized protein | 292 | 3.4247 | 0.9998 | pos: 26–27 | Plasmamembrane | 0 |
| SPSC_04676 | Uncharacterized protein | 180 | 3.3333 | 0.9997 | pos: 23–24 | Extracellular (Secreted) | 0 |
| SPSC_04678 | Uncharacterized protein | 150 | 3.3333 | 0.9998 | pos: 24–25 | Extracellular (Secreted) | 0 |
| SPSC_03601 | Uncharacterized protein | 181 | 3.3149 | 0.9997 | pos: 20–21 | Extracellular (Secreted) | 0 |
| SPSC_06259 | Uncharacterized protein | 302 | 3.3113 | 0.9998 | pos: 20–21 | Nuclear | 0 |
| SPSC_04266 | Related to Mig1 protein | 185 | 3.2432 | 0.9998 | pos: 25–26 | Extracellular (Secreted) | 0 |
| SPSC_04264 | Related to Mig1 protein | 191 | 3.1414 | 0.9998 | pos: 27–28 | Extracellular (Secreted) | 0 |
| SPSC_04267 | Related to Mig1 protein | 197 | 3.0457 | 0.9998 | pos: 27–28 | Extracellular (Secreted) | 0 |
| SPSC_04669 | Uncharacterized protein | 132 | 3.0303 | 0.9997 | pos: 24–25 | Extracellular (Secreted) | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Li, X.; Li, J.; Zhao, H.; Deng, X.; Su, Y.; Li, R.; Chen, B. Identification of Gene Modules and Hub Genes Associated with Sporisorium scitamineum Infection Using Weighted Gene Co-Expression Network Analysis. J. Fungi 2022, 8, 852. https://doi.org/10.3390/jof8080852
Liu Z, Li X, Li J, Zhao H, Deng X, Su Y, Li R, Chen B. Identification of Gene Modules and Hub Genes Associated with Sporisorium scitamineum Infection Using Weighted Gene Co-Expression Network Analysis. Journal of Fungi. 2022; 8(8):852. https://doi.org/10.3390/jof8080852
Chicago/Turabian StyleLiu, Zongling, Xiufang Li, Jie Li, Haiyun Zhao, Xingli Deng, Yizu Su, Ru Li, and Baoshan Chen. 2022. "Identification of Gene Modules and Hub Genes Associated with Sporisorium scitamineum Infection Using Weighted Gene Co-Expression Network Analysis" Journal of Fungi 8, no. 8: 852. https://doi.org/10.3390/jof8080852
APA StyleLiu, Z., Li, X., Li, J., Zhao, H., Deng, X., Su, Y., Li, R., & Chen, B. (2022). Identification of Gene Modules and Hub Genes Associated with Sporisorium scitamineum Infection Using Weighted Gene Co-Expression Network Analysis. Journal of Fungi, 8(8), 852. https://doi.org/10.3390/jof8080852