
Virulence-Associated Genes of Calonectria ilicola, Responsible for Cylindrocladium Black Rot



Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Isolation
2.2. ITS Sequencing and Phylogenetic Analysis
2.3. Inoculation of Peanut Plants with C. ilicicola
2.4. Genome Sequencing and De Novo Assembly
2.5. Gene Prediction and Annotation
2.6. RNA-Sequencing
2.7. Proteome Analysis
2.8. Bioinformatic Analysis of Differentially Expressed Proteins (DEPs)
3. Results
3.1. Identification of the C. ilicicola Ci14017 Pathogen Isolated from Peanut
3.2. Genomic Assembly and Features of C. ilicicola Ci14017
3.3. Genome and Gene Functional Annotation
3.4. Comparative Genomics Analysis
3.5. Identification of Effectors
3.6. CAZyme Analysis
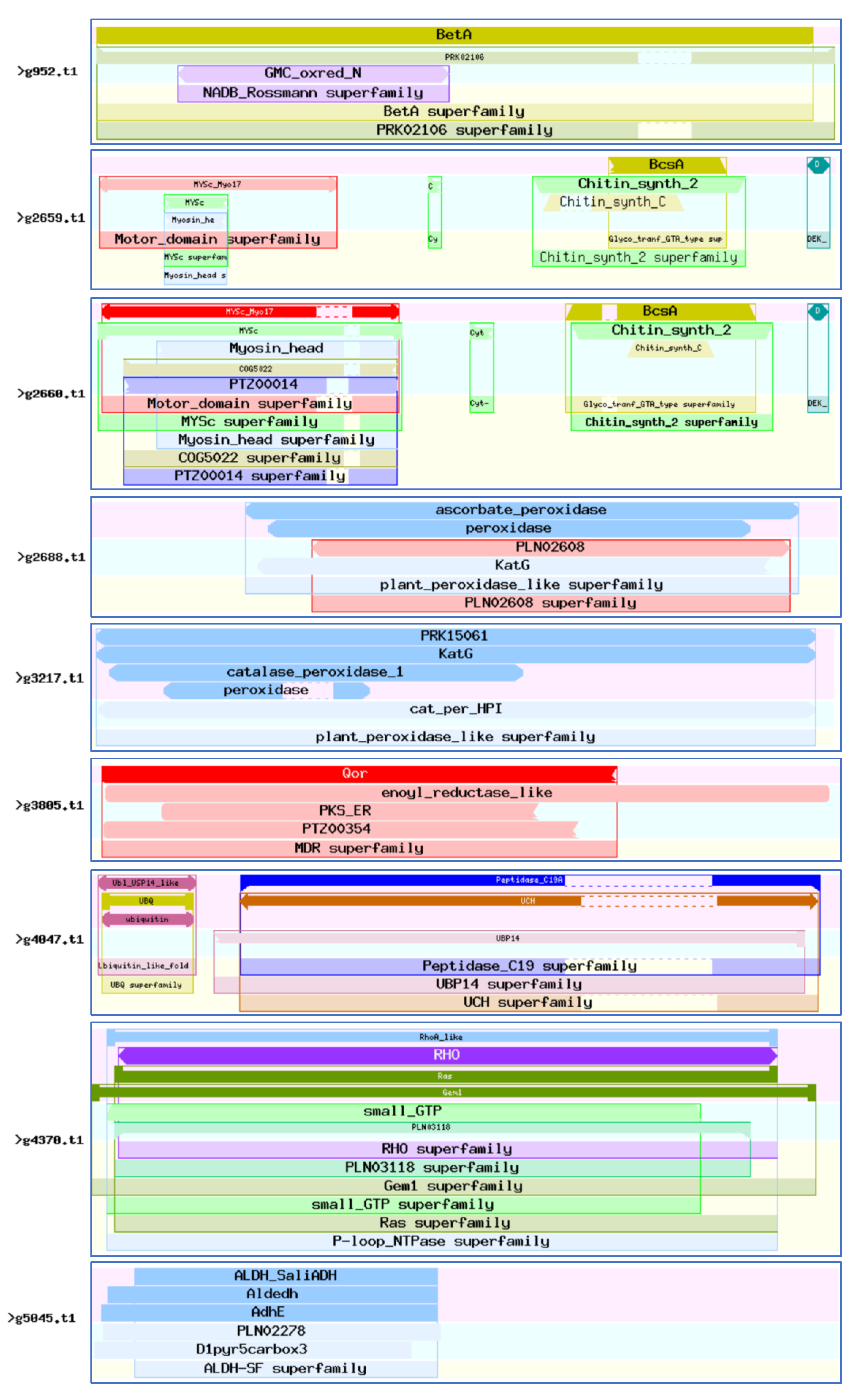
3.7. Identification of 46 Virulence-Associated Factors in C. ilicicola
3.8. Sequence Analysis of the 46 Virulence-Associated Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bell, D.K.; Sobers, E.K. A peg, pod, and root necrosis of peanuts caused by a species of Calonectria. Phytopathology 1966, 56, 1361–1364. [Google Scholar]
- Wheele, T.A.; Black, M.C. First report of Cylindrocladium black rot caused by Cylindrocladium parasiticum on peanut in Texas. Plant Dis. 2005, 89, 1245. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Wang, X.J.; Feng, L.P. Progress on the research of Cylindrocladium parasiticum. China Plant Prot. 2010, 30, 19–21. [Google Scholar]
- Tazawa, J.; Takahashi, M.; Usuki, K.; Yamamoto, H. Nodulation during vegetative growth of soybean stage does not affect the susceptibility to red crown rot caused by Calonectria ilicicloa. J. Gen. Plant Pathol. 2007, 73, 180–184. [Google Scholar] [CrossRef]
- Wright, L.P.; Davis, A.J.; Wingfield, B.D.; Crous, P.W.; Brenneman, T.; Wingfiled, M.J. Population structure of Cylindrocladium parasiticum infecting peanuts (Arachis hypogaea) in Georgia, USA. Eur. J. Plant Pathol. 2010, 127, 199–206. [Google Scholar] [CrossRef]
- Pei, W.H.; Cao, J.F.; Yang, M.Y.; Zhao, Z.J.; Xue, S.M. First report of black rot of Medicago sativa caused by Cylindrocladium parasiticum (teleomorph Calonectria ilicicola) in Yunnan Province, China. Plant Dis. 2015, 99, 890. [Google Scholar] [CrossRef]
- Fei, N.Y.; Qi, Y.B.; Meng, T.T.; Fu, J.F.; Yan, X.R. First report of root rot caused by Calonectria ilicicola on blueberry in Yunnan Province, China. Plant Dis. 2018, 102, 1036. [Google Scholar] [CrossRef]
- Zhang, Q.M.; Zhou, D.M.; Jiang, W.; Zhu, H.L.; Deng, S.; Wei, L.H. First report of soft rot of ginger caused by Calonectria ilicicola in Guangxi Province, China. Plant Dis. 2019, 104, 993. [Google Scholar] [CrossRef]
- Yi, R.H.; Su, J.J.; Li, H.J.; Li, D.; Long, G.G. First report of root rot on Manglietia decidua caused by Calonectria ilicicola in China. Plant Dis. 2022, 106, 1522. [Google Scholar] [CrossRef]
- Liu, G. List of Entry Phytosanitary Pests of the People’s Republic of China. Pestic. Mark. News 2007, 13, 38–39. [Google Scholar]
- Randall-Schadel, B.L.; Bailey, J.E.; Beute, M.K. Seed Transmission of Cylindrocladium parasiticum in Peanut. Plant Dis. 2001, 85, 362–370. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Phipps, P.M. Control of Cylindrocladium black rot of peanut with soil fumigants having methyl isothiocyanate as the active ingredient. Plant Dis. 1990, 74, 438–441. [Google Scholar] [CrossRef]
- Lan, G.B.; He, Z.F.; She, X.M.; Tang, Y.F. Identification of the resistance to black rot disease in peanut (Arachis hypogaea) cultivars caused by Cylindrocladium parasiticum in Guangdong Province, China. J. Biosaf. 2016, 25, 214–217. [Google Scholar]
- Dewey, R.E.; Siedow, J.N.; Timothy, D.H.; Levings, C.S., 3rd. A 13-kilodalton maize mitochondrial protein in E. coli confers sensitivity to Bipolaris maydis toxin. Science 1988, 239, 293–295. [Google Scholar] [CrossRef]
- Corrêa, J.A.; Hoch, H.C. Identification of thigmoresponsive loci for cell differentiation in Uromyces germlings. Protoplasma 1995, 186, 34–40. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, H.F.; Zhang, Z.G.; Zheng, X.B. GTPase activating protein MoGcs1 is important for asexual development, appressorium differentiation and stress response in the rice blast fungus Magnaporthe oryzae. Acta Phytopathol Sin. 2016, 46, 17–26. [Google Scholar]
- Yakoby, N.; Beno-Moualem, D.; Keen, N.T.; Dinoor, A.; Pines, O.; Prusky, D. Colletotrichum gloeosporioides pelB\r, is an important virulence factor in avocado fruit-fungus interaction. Mol. Plant Microbe Interact. 2001, 14, 988–995. [Google Scholar] [CrossRef]
- Gao, F.; Chu, J.M.; Li, J.H.; Wang, M.L. Research progress in the pathogenesis of plant pathogenic fungi. Jiangsu J. Agric. Sci. 2014, 30, 1174–1179. [Google Scholar]
- Wang, Y.; Chen, J.; Li, D.W.; Zheng, L.; Huang, J. CglCUT1 gene required for cutinase activity and pathogenicity of Colletotrichum gloeosporioides causing anthracnose of Camellia oleifera. Eur. J. Plant Pathol. 2017, 147, 103–114. [Google Scholar] [CrossRef]
- Shang, Y.F.; Xiao, G.H.; Zheng, P.; Cen, K.; Zhan, S.; Wang, C.S. Divergent and Convergent Evolution of Fungal Pathogenicity. Genome Biol Evol. 2016, 8, 1374–1387. [Google Scholar] [CrossRef]
- Zhang, M.L.; Xiao, S.Q.; Zhang, L.; Zhang, Y.W.; Xue, C.S. Construction deletion vector of nonribosomal peptide Synthetases 6 of Setoshpaeria turcicum. J. Maize Sci. 2012, 20, 59–63. [Google Scholar]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Wit, P.J.G.M.D.; Mehrabi, R.; Burg, H.A.V.D.; Stergiopoulos, L. Fungal effector proteins: Past, present and future. Mol. Plant Pathol. 2009, 10, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Balotf, S.; Wilson, R.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. In planta transcriptome and proteome profiles of Spongospora subterranea in resistant and susceptible host environments illuminates regulatory principles underlying host–pathogen interaction. Biology 2021, 10, 840. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.D. Isolation and culture of fungi. In Plant Disease Research Methods, 2nd ed.; Fang, Z.D., Ed.; Agriculture Press: Beijing, China, 1979; pp. 112–136. [Google Scholar]
- Skovgaard, K.; Rosendahl, S.; O’Donnell, K.; Nirenberg, H.I. Fusarium commune is a new species identified by morphological and molecular phylogenetic data. Mycologia 2003, 95, 630–636. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.D.; Lee, S.B.; Taylor, J.W. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications, 1st ed.; Innis, M.A., Ed.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Silvestro, D.; Michalak, I. raxmlGUI: A graphical front-end for RaxML. Org. Divers. Evol. 2012, 12, 335–337. [Google Scholar] [CrossRef]
- Mawhorter, R.; Libeskind-Hades, R. Hierarchical clustering of maximum parsimony reconciliations. BMC Bioinform. 2019, 20, 612. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Yao, B.; Zhang, C. DFVF: Database of fungal virulence factors. Database 2012, 2012, bas32. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, A.; Belguith, H.; Ghram, I.; Hamida, J.B. LIPABASE: A database for ’true’ lipase family enzymes. Int. J. Bioinform. Res. Appl. 2011, 7, 390. [Google Scholar] [CrossRef]
- Lisitsa, A.V.; Gusev, S.A.; Karuzina, I.I.; Archakov, A.I.; Koymans, L. Cytochrome P450 Database. SAR QSAR Environ. Res. 2001, 12, 359–366. [Google Scholar] [CrossRef]
- Winnenburg, R.; Baldwin, T.K.; Urban, M.; Rawlings, C.; Köhler, J.; Hammond-Kosack, K.E. PHI-base: A new database for pathogen host interactions. Nucleic Acids Res. 2006, 34, 459–464. [Google Scholar] [CrossRef]
- Urban, M.; Pant, R.; Raghunath, A.; Irvine, A.G.; Pedro, H.; Hammond-Kosack, K.E. The Pathogen-Host Interactions database (PHI-base): Additions and future developments. Nucleic Acids Res. 2015, 43, 645–655. [Google Scholar] [CrossRef]
- Park, B.H.; Karpinets, T.V.; Syed, M.H.; Leuze, M.R.; Uberbacher, E.C. CAZymes Analysis Toolkit (CAT): Web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology 2010, 20, 1574–1584. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; Heijne, G.V.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Nielsen, H.; Brunak, S.; Heijne, G.V. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000, 300, 1005–1016. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Brunak, S.; Heijne, G.V.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.X.; Meyer, C.A.; Wang, Q.; Liu, J.S.; Liu, X.S.; Zhang, Y. GFOLD: A generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics 2012, 28, 2782–2788. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Luo, M.; Xiang, M.M.; Xiong, L.N.; Shu, Y.X.; Yu, G.H.; Dong, Z.Y.; Sun, Y.H. Comparative proteomic analysis of the moderately resistant and susceptible peanut cultivars during infestation by Cylindrocladium parasiticum. J. Plant Pathol. 2022. submitted. [Google Scholar]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar]
- Liu, H.H.; Wang, J.; Wu, P.H.; Lu, M.Y.; Li, J.Y.; Shen, Y.M.; Tzeng, M.N.; Kuo, C.H.; Lin, Y.H.; Chang, H.X. Whole-Genome Sequence Resource of Calonectria ilicicola, the Casual Pathogen of Soybean Red Crown Rot. Mol. Plant Microbe Interact. 2021, 34, 848–851. [Google Scholar] [CrossRef]
- Cao, C.J.; Xue, C.Y. More Than Just Cleaning: Ubiquitin-Mediated Proteolysis in Fungal Pathogenesis. Front Cell Infect. Microbiol. 2021, 11, 774613. [Google Scholar] [CrossRef]
- Liu, T.B.; Xue, C.Y. The Ubiquitin-Proteasome System and F-box Proteins in Pathogenic Fungi. Mycobiology 2011, 39, 243–248. [Google Scholar] [CrossRef]
- Han, Y.K.; Kim, M.D.; Lee, S.H.; Yun, S.H.; Lee, Y.W. A Novel F-Box Protein Involved in Sexual Development and Pathogenesis in Gibberella zeae. Mol. Microbiol. 2007, 63, 768–779. [Google Scholar] [CrossRef]
- Bartnicki-Garcia, S.; Bartnicki, D.D.; Gierz, G. Determinants of fungal cell-wall morphology: The vesicle supply center. Can. J. Bot. 1995, 73, 372–378. [Google Scholar] [CrossRef]
- Werner, S.; Sugui, J.A.; Steinberg, G.; Deising, H.B. A chitin synthase with a myosin-like motor domain is essential for hyphal growth, appressorium differentiation, and pathogenicity of the maize anthracnose fungus Colletotrichum graminicola. Mol. Plant Microbe Interact. 2007, 20, 1555–1567. [Google Scholar] [CrossRef]
- Zhang, J.J.; Jiang, H.; Du, Y.R.; Keyhani, N.O.; Xia, Y.X.; Jin, K. Members of chitin synthase family in Metarhizium acridum differentially affect fungal growth, stress tolerances, cell wall integrity and virulence. PLoS Pathog. 2019, 15, e1007964. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Koymans, L.; Kamataki, T.; Stegeman, J.J.; Feyereisen, R.; Waxman, D.J.; Waterman, M.R.; Gotoh, O.; Coon, M.J.; Estabrook, R.W.; et al. P450 superfamily: Update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 1996, 6, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Brink, H.M.V.D.; Gorcom, R.F.V.; Hondel, C.A.V.D.; Punt, P.J. Cytochrome P450 enzyme systems in fungi. Fungal Genet. Biol. 1998, 23, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Črešnar, B.; Petrič, Š. Cytochrome P450 enzymes in the fungal kingdom. Biochim. Biophys. Acta 2011, 1814, 29–35. [Google Scholar] [CrossRef]
- Shin, J.Y.; Bui, D.C.; Lee, Y.; Nam, H.J.; Jung, S.Y.; Fang, M.; Kim, J.; Lee, T.; Kim, H.; Choi, G.J.; et al. Functional characterization of cytochrome P450 monooxygenases in the cereal head blight fungus Fusarium graminearum. Environ. Microbiol. 2017, 19, 2053–2067. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.Y.; Chai, R.Y.; Qiu, H.P.; Jiang, H.; Mao, X.Q.; Wang, Y.L.; Liu, F.Q.; Sun, G.C. An S-(Hydroxymethyl) glutathione dehydrogenase is involved in conidiation and full virulence in the rice blast fungus Magnaporthe oryzae. PLoS ONE 2015, 10, e0120627. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ci14017 | Statistical Data |
|---|---|
| The number of scaffolds | 1993 |
| Assembly size (bp) | 66,310,315 |
| Mean length (bp) | 33,272 |
| Max length (bp) | 1,935,007 |
| Min length (bp) | 121 |
| Largest 10 total (bp) | 14,067,989 |
| The number of scaffolds (>1000 bp) | 820 |
| Scaffold N50 (bp) | 390,247 |
| Number of gaps | 442 |
| Total gap length (bp) | 244,347 |
| Mean gap length (bp) | 553 |
| Percent N’s | 0.37% |
| GC content | 48% |
| Repetitive DNA | 1.53% |
| tRNA | 423 |
| rRNA | 90 |
| Predicted coding genes | 18,366 |
| Total of exons | 53,680 |
| Coding region in genome | 42.23% |
| Mean gene length (bp) | 3610 |
| Mean coding gene length (bp) | 1525 |
| Mean exons per gene | 3 |
| Mean exon length (bp) | 522 |
| SeqName | Description | Annotation to Categorize | Transcriptome/Fold Change (PC/TC) | Proteome/Fold Change (PC/TC) | Length |
|---|---|---|---|---|---|
| g11138.t1 | Cell division control protein 48 | Lipase/P450 | 0.645397155 | 3.817191977 | 836 |
| g11761.t1 | CVNH domain-containing protein | CAZymes | 1.077780219 | 2.576375314 | 409 |
| g12183.t1 | NADPH-cytochrome P450 reductase | Lipase/P450 | 0.777423319 | 2.274784162 | 690 |
| g12364.t1 | Peptidyl-prolyl cis-trans isomerase | / | 0.7462787 | 2.408002106 | 114 |
| g13131.t1 | alcohol dehydrogenase I | Lipase | 0.850951116 | 4.617233843 | 350 |
| g13442.t1 | Apoptosis-inducing factor 1 | P450 | 3.084508684 | 3.559369202 | 546 |
| g13926.t1 | Acetyl-coenzyme A synthetase | Lipase/P450 | 2.340024168 | 2.520666667 | 704 |
| g14087.t1 | S-(hydroxymethyl)glutathione dehydrogenase | Lipase | 0.35893699 | 3.679765396 | 888 |
| g14473.t1 | Fatty acid synthase subunit alpha | P450 | 1.920882975 | 2.612333333 | 1860 |
| g15360.t1 | 1,3-beta-glucanosyltransferase Gel1 | CAZymes | 1.406499686 | 2.461296413 | 453 |
| g15638.t1 | T-complex protein 1 subunit eta | Lipase | 0.933011105 | 2 | 557 |
| g15744.t1 | Glutamate decarboxylase | P450 | 0.89437205 | 3.28017126 | 571 |
| g15844.t1 | eIF4A-like protein | Lipase/P450 | 0.395142551 | 2.8717477 | 397 |
| g15879.t1 | CAMK/CAMK1/CAMK1-CMK protein kinase | Lipase/P450 | 1.036268466 | 2.70942813 | 370 |
| g15902.t1 | Carboxypeptidase Y A | Lipase/P450/Merops | 1.822742632 | 2.263530052 | 544 |
| g15950.t1 | Actin cytoskeleton-regulatory complex protein PAN1 | P450 | 0.418771014 | 1.666666667 | 1501 |
| g16158.t1 | Glucooligosaccharide oxidase | Lipase/P450/CAZymes/Secrete | 0.375885577 | 2.000333333 | 518 |
| g16583.t1 | endopolygalacturonase 1 | CAZymes/Secrete | 0.128246495 | 2.894074074 | 359 |
| g16828.t1 | probable endopolygalacturonase NFIA_008150 | CAZymes/Secrete | 0.758522499 | 1.909192825 | 382 |
| g17377.t1 | Thioredoxin reductase | Lipase | 0.46674335 | 2.260225252 | 321 |
| g18104.t1 | ubiquitin-conjugating enzyme E2-16 kDa | P450 | 0.804791205 | 1.666666667 | 146 |
| g18127.t1 | vacuolar protease A | P450/Merops | 1.864883004 | 2.301609848 | 502 |
| g18177.t1 | vesicle fusion factor | Lipase/P450 | 0.489926572 | 2.182333333 | 849 |
| g18328.t1 | elongation factor 3 | Lipase/P450 | 0.708137425 | 3.309420682 | 1055 |
| g18352.t1 | succinate-semialdehyde dehydrogenase (NADP+) | Lipase/P450 | 0.237659648 | 3.404154863 | 494 |
| g2659.t1 | chitin synthase | CAZymes | 1.141364798 | 1.666666667 | 1773 |
| g2660.t1 | chitin synthase | CAZymes | 1.075445163 | 1.999666667 | 1857 |
| g2688.t1 | Cytochrome c peroxidase, mitochondrial | CAZymes | 1.381410476 | 2.560020346 | 357 |
| g3217.t1 | catalase-peroxidase | CAZymes | 0.853030366 | 2.82071459 | 762 |
| g3305.t1 | NAD-binding Rossmann fold oxidoreductase family protein | CAZymes | 262.7 | 2 | 440 |
| g3805.t1 | GroES-like protein | Lipase/P450 | 0.450581519 | 2.705308465 | 357 |
| g4047.t1 | ubiquitin thiolesterase | Merops | 1.087713026 | 2 | 555 |
| g4370.t1 | GTP-binding protein rhoA | Lipase | 1.469419616 | 2.352234637 | 195 |
| g5045.t1 | Vanillin dehydrogenase | Lipase/P450 | 0.73931864 | 2.946907653 | 1021 |
| g5049.t1 | related to short chain dehydrogenase | Lipase/P450 | 0.908852984 | 2.114715806 | 258 |
| g5051.t1 | related to 6-hydroxy-D-nicotine oxidase | Lipase/P450/CAZymes | 0.727663175 | 2.707425743 | 461 |
| g5191.t1 | nitric oxide dioxygenase | P450 | 0.177916187 | 2 | 420 |
| g5581.t1 | Cytochrome P450 55A3 | Lipase/P450 | 0.595411287 | 1.999666667 | 404 |
| g5899.t1 | fungal specific transcription factor | Lipase/CAZymes/Secrete | 0.148888017 | 2.146453089 | 1191 |
| g6789.t1 | cysteine desulfurase | Lipase | 1.597153312 | 2 | 504 |
| g7130.t1 | glucosamine-fructose-6-phosphate aminotransferase | Merops | 0.508395005 | 3.672040022 | 702 |
| g7426.t1 | t-complex protein 1 subunit beta | Lipase | 0.053649734 | 2.256364713 | 540 |
| g7739.t1 | Peptidoglycan deacetylase | CAZymes | 43.97 | 1.666666667 | 335 |
| g7873.t1 | S-adenosyl-L-methionine-dependent methyltransferase | Lipase | 37.26 | 2 | 397 |
| g8661.t1 | predicted protein | P450 | 0.499370829 | 1.999666667 | 886 |
| g952.t1 | alcohol oxidase | P450/CAZymes | 0.340802231 | 3.471797575 | 688 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Luo, M.; Wu, W.; Dong, Z.; Zou, H. Virulence-Associated Genes of Calonectria ilicola, Responsible for Cylindrocladium Black Rot. J. Fungi 2022, 8, 869. https://doi.org/10.3390/jof8080869
Chen X, Luo M, Wu W, Dong Z, Zou H. Virulence-Associated Genes of Calonectria ilicola, Responsible for Cylindrocladium Black Rot. Journal of Fungi. 2022; 8(8):869. https://doi.org/10.3390/jof8080869
Chicago/Turabian StyleChen, Xinyu, Mei Luo, Wei Wu, Zhangyong Dong, and Huasong Zou. 2022. "Virulence-Associated Genes of Calonectria ilicola, Responsible for Cylindrocladium Black Rot" Journal of Fungi 8, no. 8: 869. https://doi.org/10.3390/jof8080869
APA StyleChen, X., Luo, M., Wu, W., Dong, Z., & Zou, H. (2022). Virulence-Associated Genes of Calonectria ilicola, Responsible for Cylindrocladium Black Rot. Journal of Fungi, 8(8), 869. https://doi.org/10.3390/jof8080869