
Soil Layers Impact Lithocarpus Soil Microbial Composition in the Ailao Mountains Subtropical Forest, Yunnan, China

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Description of the Sampling Site
2.2. Experimental Design and Sample Collection
2.3. DNA Extraction, PCR Amplification, and Sequencing
2.4. Processing and Analyses of the Sequencing Data
2.5. Statistical Analysis and Visualization
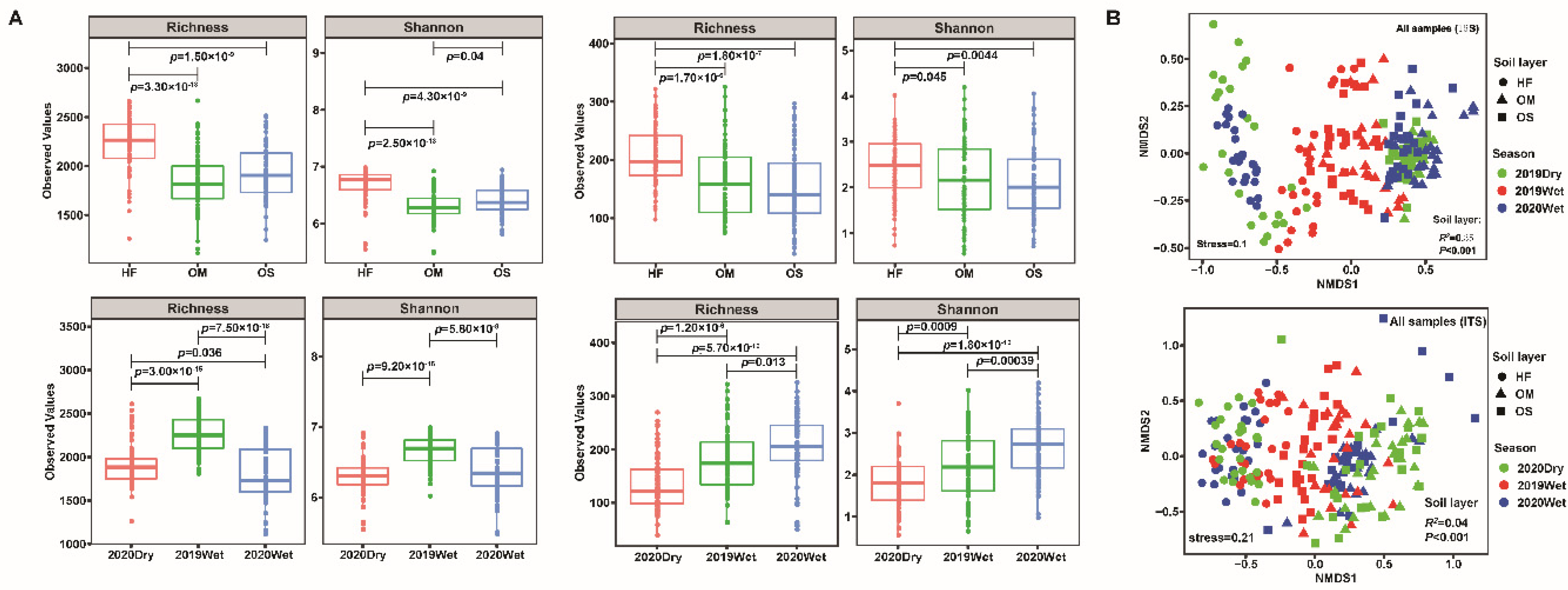
3. Results
3.1. Soil Microbiome Assembly Was Most Strongly Influenced by the Soil Layer
3.2. Soil Layers Possessed Different Ecological Network Complexity
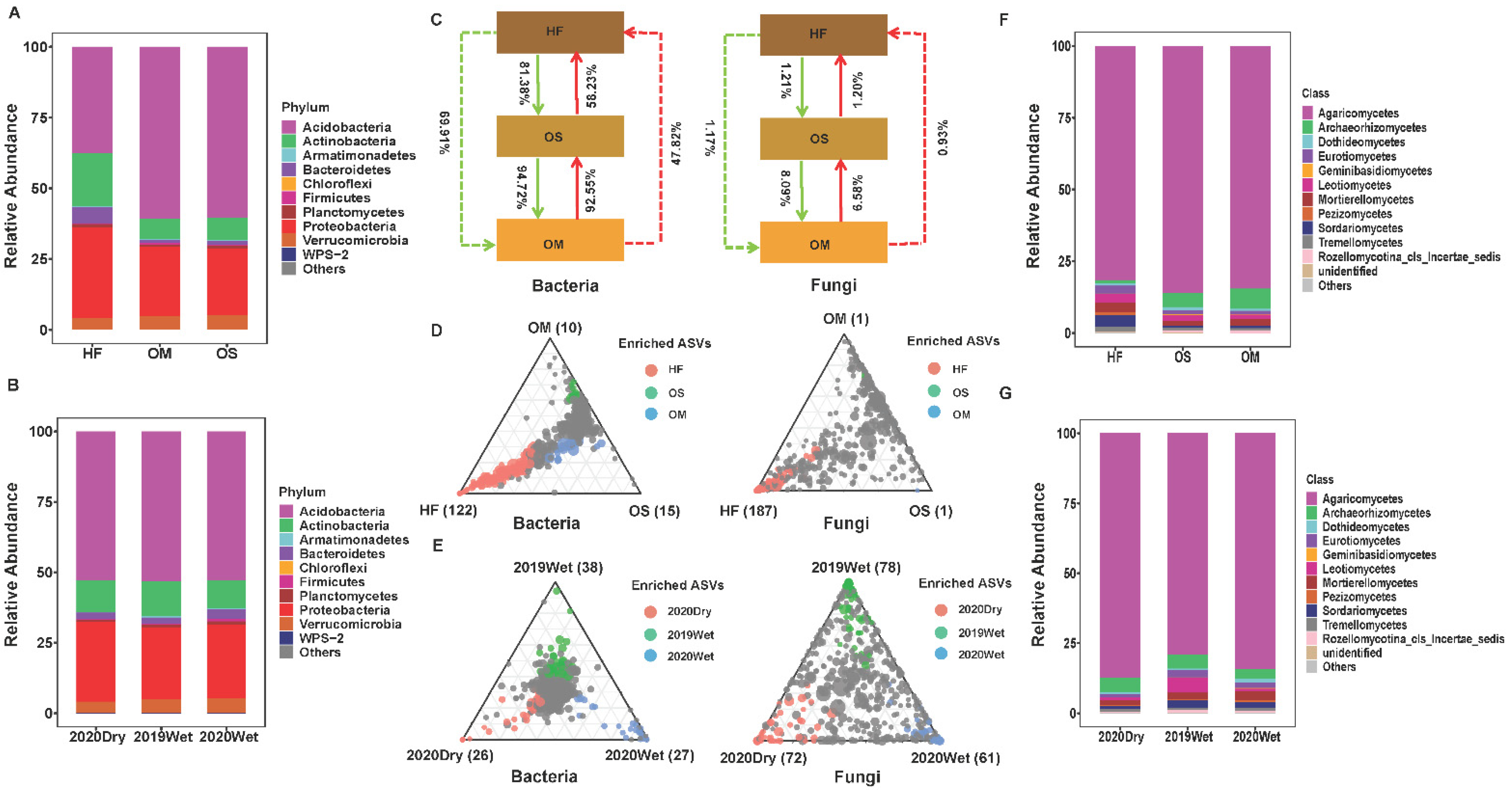
3.3. The Community Composition and Selection Process of Soil Layer
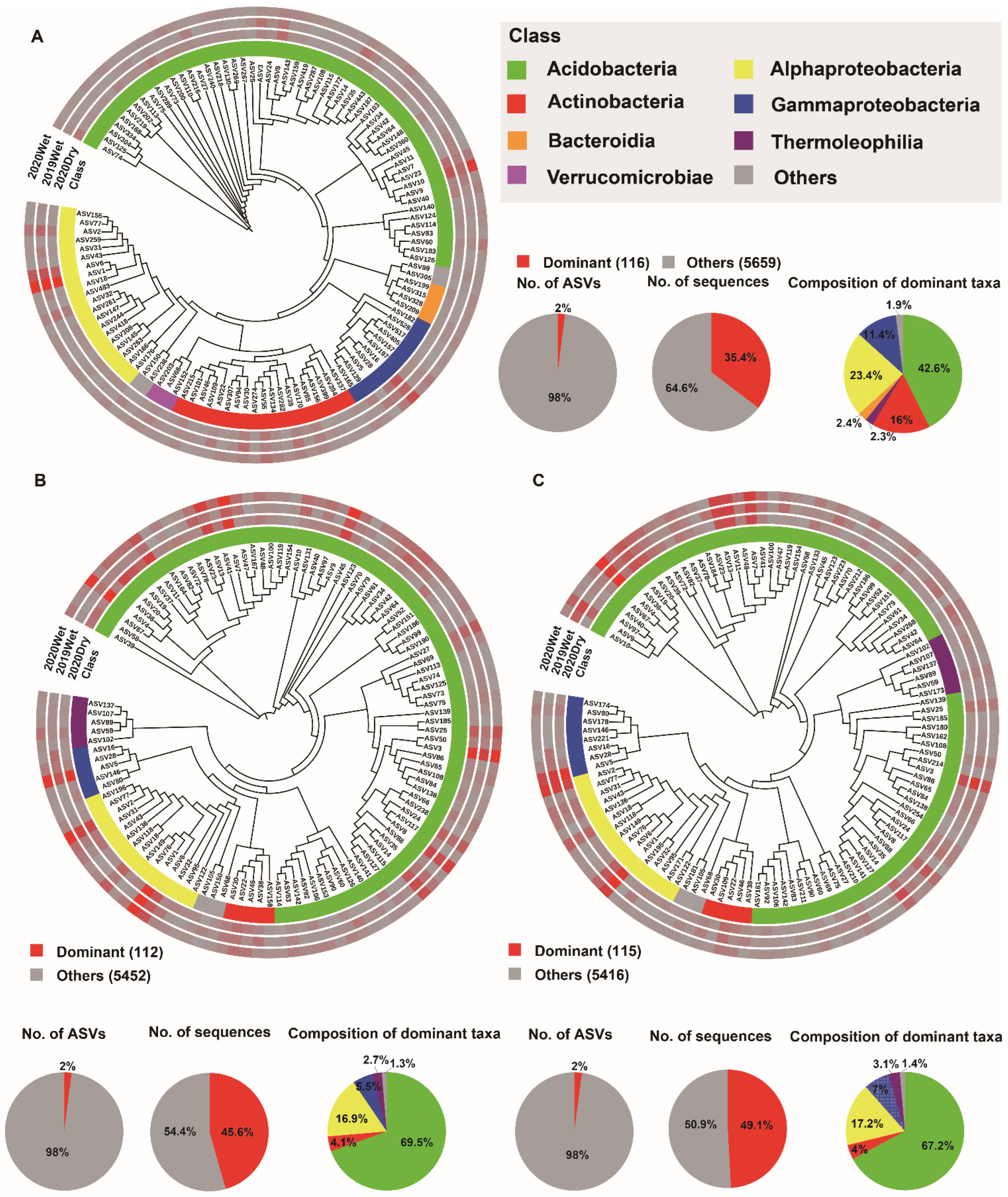
3.4. Soil Microbiome Dominant Taxa and Biomarker among Soil Layers
4. Discussion
4.1. The Assembly Pattern and Divergence of Soil Layer Microbiome
4.2. The Community Composition and Selection Process of Soil Layer
4.3. The Dominant Taxa for Each Soil Layer
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Petr, B. Forest microbiome: Diversity, complexity and dynamics. FEMS Microbiol. Rev. 2016, 41, 109–130. [Google Scholar]
- Mishra, S.; Httenschwiler, S.; Yang, X. The plant microbiome: A missing link for the understanding of community dynamics and multifunctionality in forest ecosystems. Appl. Soil Ecol. 2020, 145, 103345. [Google Scholar] [CrossRef]
- Thakur, M.P.; Geisen, S. Trophic Regulations of the Soil Microbiome. Trends Microbiol. 2019, 27, 771–780. [Google Scholar] [CrossRef]
- Wagg, C.; Schlaeppi, K.; Banerjee, S.; Kuramae, E.E.; van der Heijden, M.G.A. Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nat. Commun. 2019, 10, 4841. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.L.; Ding, J.; Zhu, Y.G.; He, J.Z.; Hu, H.W. Soil bacterial taxonomic diversity is critical to maintaining the plant productivity. Environ. Int. 2020, 140, 105766. [Google Scholar] [CrossRef]
- Van der Putten, W.H.; Bardgett, R.D. Belowground biodiversity and ecosystem functioning. Nature 2014, 515, 505–511. [Google Scholar]
- Tao, C.; Li, R.; Xiong, W.; Shen, Z.; Liu, S.; Wang, B.; Ruan, Y.; Geisen, S.; Shen, Q.; Kowalchuk, G.A. Bio-organic fertilizers stimulate indigenous soil Pseudomonas populations to enhance plant disease suppression. Microbiome 2020, 8, 137. [Google Scholar] [CrossRef]
- Veach, A.M.; Morris, R.; Yip, D.Z.; Yang, Z.K.; Engle, N.L.; Cregger, M.A.; Tschaplinski, T.J.; Schadt, C.W. Correction to: Rhizosphere microbiomes diverge among Populus trichocarpa plant-host genotypes and chemotypes, but it depends on soil origin. Microbiome 2021, 9, 1–2. [Google Scholar] [CrossRef]
- Zhuang, L.; Li, Y.; Wang, Z.; Yu, Y.; Zhang, N.; Yang, C.; Zeng, Q.; Wang, Q. Synthetic community with six Pseudomonas strains screened from garlic rhizosphere microbiome promotes plant growth. Microb. Biotechnol. 2021, 14, 488–502. [Google Scholar] [CrossRef]
- Bettenfeld, P.; Fontaine, F.; Trouvelot, S.; Fernandez, O.; Courty, P.E. Woody Plant Declines. What’s Wrong with the Microbiome? Trends Plant Sci. 2020, 25, 381–394. [Google Scholar] [CrossRef]
- Beckers, B.; Beeck, M.O.D.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; He, J.Z.; Singh, B.K.; Zhu, Y.G.; Zhang, L. Rare taxa maintain the stability of crop mycobiomes and ecosystem functions. Environ. Microbiol. 2020, 23, 1907–1924. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Jaramillo, J.E.; Carrión, V.J.; Bosse, M.; Ferrão, L.F.V.; de Hollander, M.; Garcia, A.A.F.; Ramírez, A.C.; Mendes, R.; Raaijmakers, J.M. Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits. ISME J. 2017, 11, 2244–2257. [Google Scholar] [CrossRef]
- Bonito, G.; Reynolds, H.; Robeson, M.S.; Nelson, J.; Hodkinson, B.P.; Tuskan, G.; Schadt, C.W.; Vilgalys, R. Plant host and soil origin influence fungal and bacterial assemblages in the roots of woody plants. Mol. Ecol. 2014, 23, 3356–3370. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.J.; Wang, J.M.; Bai, W.Q.; Qi, J.J.; Chen, W.M. Soil bacterial diversity correlates with precipitation and soil pH in long-term maize cropping systems. Sci. Rep. 2020, 10, 6012. [Google Scholar] [CrossRef]
- Gao, Z.; Han, M.; Hu, Y.; Li, Z.; Ma, Z. Effects of Continuous Cropping of Sweet Potato on the Fungal Community Structure in Rhizospheric Soil. Front. Microbiol. 2019, 10, 2269. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, W.; Wang, J.; Du, N.; Li, Q.; Wei, G. Soil microbiomes with distinct assemblies through vertical soil profiles drive the cycling of multiple nutrients in reforested ecosystems. Microbiome 2018, 6, 146. [Google Scholar] [CrossRef]
- Du, X.; Deng, Y.; Li, S.; Escalas, A.; Feng, K.; He, Q.; Wang, Z.; Wu, Y.; Wang, D.; Peng, X.; et al. Steeper spatial scaling patterns of subsoil microbiota are shaped by deterministic assembly process. Mol. Ecol. 2021, 30, 1072–1085. [Google Scholar] [CrossRef]
- Fierer, N.; Schimel, J.P.; Holden, P.A. Variations in microbial community composition through two soil depth profiles. Soil Biol. Biochem. 2003, 35, 167–176. [Google Scholar] [CrossRef]
- Eichlerová, I.; Homolka, L.; Žifčáková, L.; Lisá, L.; Dobiášová, P.; Baldrian, P. Enzymatic systems involved in decomposition reflects the ecology and taxonomy of saprotrophic fungi. Fungal Ecol. 2015, 13, 10–22. [Google Scholar] [CrossRef]
- López-Mondéjar, R.; Voříšková, J.; Větrovsky, T.; Baldrian, P. The bacterial community inhabiting temperate deciduous forests is vertically stratified and undergoes seasonal dynamics. Soil Biol. Biochem. 2015, 87, 43–50. [Google Scholar] [CrossRef]
- Strickland, M.S.; Rousk, J. Considering fungal:bacterial dominance in soils—Methods, controls, and ecosystem implications. Soil Biol. Biochem. 2010, 42, 1385–1395. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, L.; Luo, Y.; Awasthi, M.K.; Yang, J.; Duan, Y.; Li, H.; Zhao, Z. Mulching practices alter the bacterial-fungal community and network in favor of soil quality in a semiarid orchard system. Sci. Total Environ. 2020, 725, 138527. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhao, Z.; Gong, Q.; Zhai, B.; Li, Z. Responses of fungal–bacterial community and network to organic inputs vary among different spatial habitats in soil. Soil Biol. Biochem. 2018, 125, 54–63. [Google Scholar] [CrossRef]
- Ashton, L.A.; Nakamura, A.; Burwell, C.J.; Tang, Y.; Cao, M.; Whitaker, T.; Sun, Z.; Huang, H.; Kitching, R.L. Elevational sensitivity in an Asian ‘hotspot’: Moth diversity across elevational gradients in tropical, sub-tropical and sub-alpine China. Sci. Rep. 2016, 6, 26513. [Google Scholar] [CrossRef]
- Young, S.S.; Carpenter, C.; Wang, Z.J. A study of the structure and composition of an old growth and secondary broad-leaved forest in the Ailao mountains of Yunnan, China. Mt. Res. Dev. 1992, 12, 269–284. [Google Scholar] [CrossRef]
- Song, Q.H.; Fei, X.H.; Zhang, Y.P.; Sha, L.Q.; Liu, Y.T.; Zhou, W.J.; Wu, C.S.; Lu, Z.Y.; Luo, K.; Gao, J.B.; et al. Water use efficiency in a primary subtropical evergreen forest in Southwest China. Sci. Rep. 2017, 7, 43031. [Google Scholar] [CrossRef]
- Šnajdr, J.; Valášková, V.; Merhautová, V.; Herinková, J.; Cajthaml, T.; Baldrian, P. Spatial variability of enzyme activities and microbial biomass in the upper layers of Quercus petraea forest soil. Soil Biol. Biochem. 2008, 40, 2068–2075. [Google Scholar] [CrossRef]
- Lindahl, B.D.; Ihrmark, K.; Boberg, J.; Trumbore, S.E.; Högberg, P.; Stenlid, J.; Finlay, R.D. Spatial separation of litter decomposition and mycorrhizal nitrogen uptake in a boreal forest. New Phytol. 2007, 173, 611–620. [Google Scholar] [CrossRef]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes—Application to the identification of mycorrhizae and rusts. Mol. Ecol. 2010, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcmurdie, P.J.; Susan, H.; Michael, W. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Zhu, Y.-G.; Wang, J.-T.; Singh, B.; Han, L.-L.; Shen, J.-P.; Li, P.-P.; Wang, G.-B.; Wu, C.-F.; Ge, A.-H.; et al. Host selection shapes crop microbiome assembly and network complexity. New Phytol. 2021, 229, 1091–1104. [Google Scholar] [CrossRef]
- Dan, K.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar]
- Kurtz, Z.D.; Mueller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLOS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef]
- Liu, H.; Roeder, K.; Wasserman, L. Stability Approach to Regularization Selection (StARS) for High Dimensional Graphical Models. Adv. Neural Inf. Process. Syst. 2010, 24, 1432–1440. [Google Scholar]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the Third International Conference on Weblogs and Social Media, ICWSM 2009, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Hamilton, N.E.; Ferry, M. ggtern: Ternary Diagrams Using ggplot2. J. Stat. Softw. 2018, 87, 1–17. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Eilers, K.G.; Debenport, S.; Anderson, S.; Fierer, N. Digging deeper to find unique microbial communities: The strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol. Biochem. 2012, 50, 58–65. [Google Scholar] [CrossRef]
- Wu, H.; Adams, J.M.; Shi, Y.; Li, Y.; Song, X.; Zhao, X.; Chu, H.; Zhang, G.L. Depth-Dependent Patterns of Bacterial Communities and Assembly Processes in a Typical Red Soil Critical Zone. Geomicrobiol. J. 2020, 37, 201–212. [Google Scholar] [CrossRef]
- Zifcakova, L.; Vetrovsky, T.; Howe, A.; Baldrian, P. Microbial activity in forest soil reflects the changes in ecosystem properties between summer and winter. Environ. Microbiol. 2016, 18, 288–301. [Google Scholar] [CrossRef] [Green Version]
- Oita, S.; Ibáez, A.; Lutzoni, F.; Miadlikowska, J.; Geml, J.; Lewis, L.A.; Hom, E.F.Y.; Carbone, I.; U’Ren, J.M.; Arnold, A.E. Climate and seasonality drive the richness and composition of tropical fungal endophytes at a landscape scale. Commun. Biol. 2021, 4, 313. [Google Scholar] [CrossRef]
- Schimel, J.P.; Schaeffer, S.M. Microbial control over carbon cycling in soil. Front. Microbiol. 2012, 3, 348. [Google Scholar] [CrossRef]
- Cordero, O.X.; Datta, M.S. Microbial interactions and community assembly at microscales. Curr. Opin. Microbiol. 2016, 31, 227–234. [Google Scholar] [CrossRef]
- De Vries, F.T.; Griffiths, R.I.; Bailey, M.; Craig, H.; Girlanda, M.; Gweon, H.S.; Hallin, S.; Kaisermann, A.; Keith, A.M.; Kretzschmar, M. Soil bacterial networks are less stable under drought than fungal networks. Nat. Commun. 2018, 9, 3033. [Google Scholar] [CrossRef]
- Leach, J.E.; Triplett, L.R.; Argueso, C.T.; Trivedi, P. Communication in the Phytobiome. Cell 2017, 169, 587. [Google Scholar] [CrossRef]
- Berg, G.; Smalla, K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol. Ecol. 2009, 68, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vorholt, J.A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 2012, 10, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Remus-Emsermann, M.N.P.; Schlechter, R.O. Phyllosphere microbiology: At the interface between microbial individuals and the plant host. New Phytol. 2018, 218, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Hamonts, K.; Trivedi, P.; Garg, A.; Janitz, C.; Grinyer, J.; Holford, P.; Botha, F.C.; Anderson, I.C.; Singh, B.K. Field study reveals core plant microbiota and relative importance of their drivers. Environ. Microbiol. 2018, 20, 124–140. [Google Scholar] [CrossRef]
- Hu, Q.; Tan, L.; Gu, S.; Xiao, Y.; Xiong, X.; Zeng, W.-a.; Feng, K.; Wei, Z.; Deng, Y. Network analysis infers the wilt pathogen invasion associated with non-detrimental bacteria. npj Biofilms Microbiomes 2020, 6, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Tedersoo, L.; Bahram, M.; Polme, S.; Anslan, S.; Riit, T.; Koljalg, U.; Nilsson, R.H.; Hildebrand, F.; Abarenkov, K. Response to Comment on “Global diversity and geography of soil fungi”. Science 2015, 349, 936. [Google Scholar] [CrossRef]
- Coller, E.; Cestaro, A.; Zanzotti, R.; Bertoldi, D.; Donati, C. Microbiome of vineyard soils is shaped by geography and management. Microbiome 2019, 7, 140. [Google Scholar] [CrossRef]
- Fontaine, S.; Barot, S.; Barré, P.; Bdioui, N.; Mary, B.; Rumpel, C. Stability of organic carbon in deep soil layers controlled by fresh carbon supply. Nature 2007, 450, 277–280. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-Gonzalez, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Kalam, S.; Basu, A.; Ahmad, I.; Sayyed, R.Z.; Suriani, N.L. Recent Understanding of Soil Acidobacteria and Their Ecological Significance: A Critical Review. Front. Microbiol. 2020, 11, 580024. [Google Scholar] [CrossRef] [PubMed]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; van der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Sung, G.-H.; López-Giráldez, F.; Townsend, J.P.; Miadlikowska, J.; Hofstetter, V.; Robbertse, B.; Matheny, P.B.; Kauff, F.; Wang, Z.; et al. The Ascomycota Tree of Life: A Phylum-wide Phylogeny Clarifies the Origin and Evolution of Fundamental Reproductive and Ecological Traits. Syst. Biol. 2009, 58, 224–239. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil Layer | Node | Positive Edge | Negative Edge | Average Degree | Modularity | Average Clustering Coefficient | Average Path Distance | Hub Node | |
|---|---|---|---|---|---|---|---|---|---|
| Bacteria | HF | 308 | 977 | 571 | 10.05 | 1.561 | 0.454 | 3.115 | 23 |
| OS | 439 | 1503 | 985 | 11.34 | 1.905 | 0.335 | 3.143 | 43 | |
| OM | 444 | 1246 | 691 | 8.73 | 1.449 | 0.324 | 3.361 | 20 | |
| Fungi | HF | 512 | 1605 | 220 | 7.13 | 0.815 | 0.355 | 4.063 | 24 |
| OS | 357 | 994 | 100 | 6.13 | 0.799 | 0.349 | 3.902 | 6 | |
| OM | 353 | 857 | 97 | 5.41 | 0.82 | 0.364 | 3.903 | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Wu, J.; Wang, H.; Lukianova, A.; Tokmakova, A.; Jin, Z.; Tan, S.; Chen, S.; Wang, Y.; Du, Y.; et al. Soil Layers Impact Lithocarpus Soil Microbial Composition in the Ailao Mountains Subtropical Forest, Yunnan, China. J. Fungi 2022, 8, 948. https://doi.org/10.3390/jof8090948
Liu S, Wu J, Wang H, Lukianova A, Tokmakova A, Jin Z, Tan S, Chen S, Wang Y, Du Y, et al. Soil Layers Impact Lithocarpus Soil Microbial Composition in the Ailao Mountains Subtropical Forest, Yunnan, China. Journal of Fungi. 2022; 8(9):948. https://doi.org/10.3390/jof8090948
Chicago/Turabian StyleLiu, Sijia, Jiadong Wu, Haofei Wang, Anna Lukianova, Anna Tokmakova, Zhelun Jin, Shuxian Tan, Sisi Chen, Yue Wang, Yuxin Du, and et al. 2022. "Soil Layers Impact Lithocarpus Soil Microbial Composition in the Ailao Mountains Subtropical Forest, Yunnan, China" Journal of Fungi 8, no. 9: 948. https://doi.org/10.3390/jof8090948
APA StyleLiu, S., Wu, J., Wang, H., Lukianova, A., Tokmakova, A., Jin, Z., Tan, S., Chen, S., Wang, Y., Du, Y., Miroshnikov, K. A., & Xie, J. (2022). Soil Layers Impact Lithocarpus Soil Microbial Composition in the Ailao Mountains Subtropical Forest, Yunnan, China. Journal of Fungi, 8(9), 948. https://doi.org/10.3390/jof8090948