
Integrated Transcriptome and Untargeted Metabolomic Analyses Revealed the Role of Methyltransferase Lae1 in the Regulation of Phospholipid Metabolism in Trichoderma atroviride




Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strains and Growth Conditions
2.2. Total RNA Extraction and RNA Sequencing
2.3. Identification of DEGs and Functional Enrichment Analyses
2.4. Extraction of Metabolites
2.5. UPLC-QTOF-MS/MS Analysis
2.6. Multivariate Statistical Analysis
2.7. Differential Metabolite Analysis
3. Results
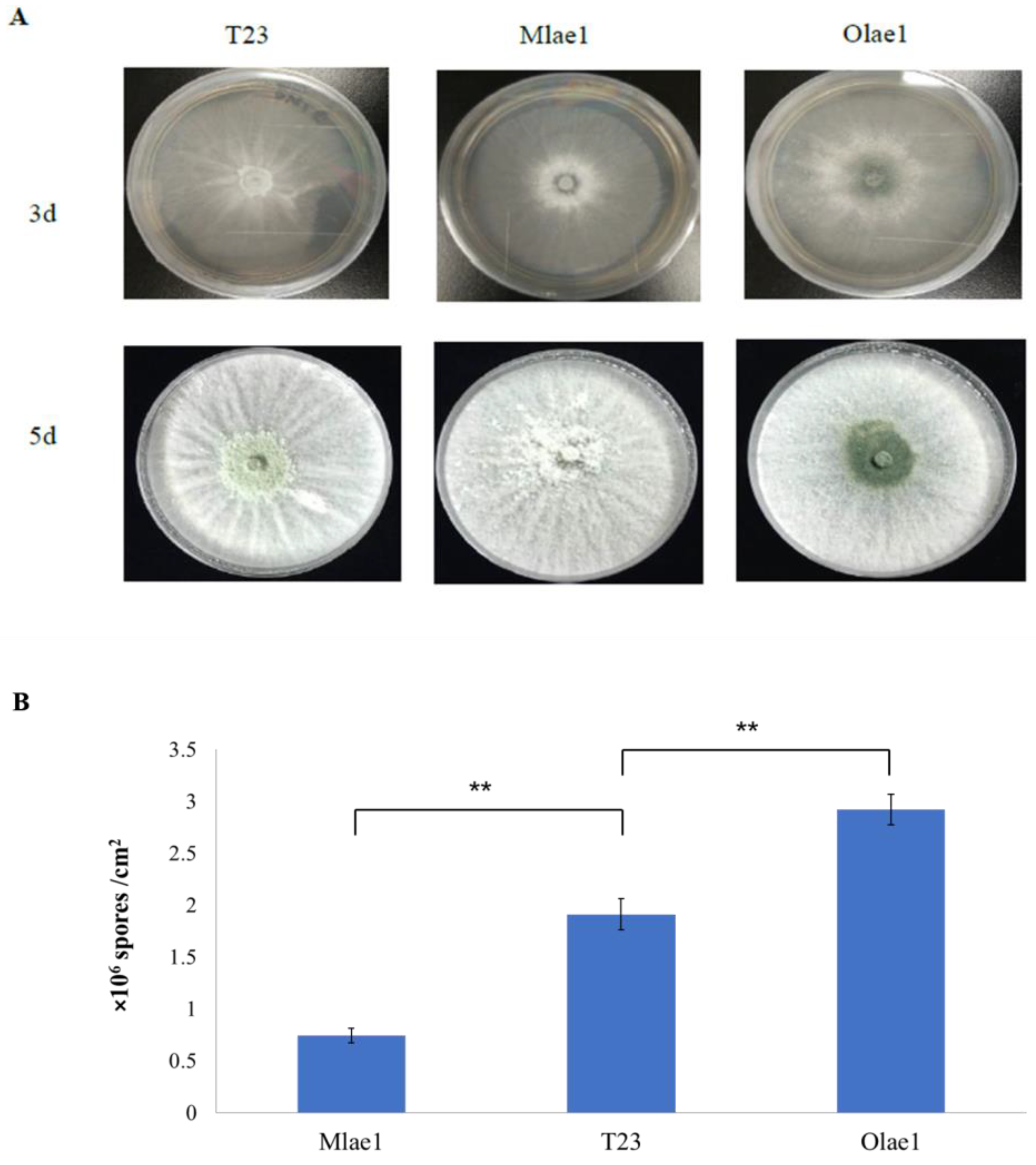
3.1. Effects of Lae1 on Phenotype in Trichoderma atroviride 23
3.2. Profiling the Transcriptome of Three Strains: Analysis of Differentially Expressed Genes (DEGs)
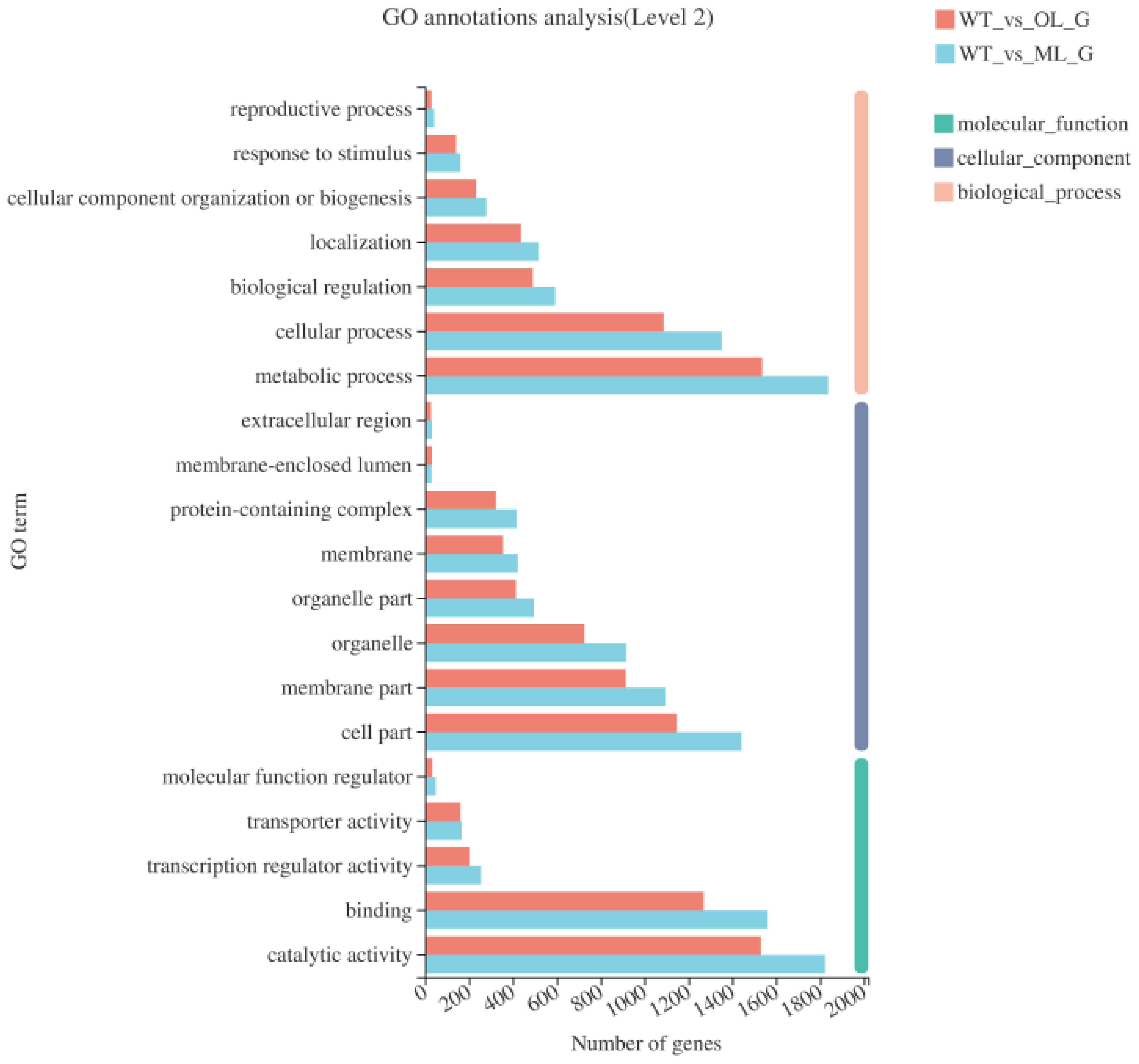
3.3. GO Functional Analysis of Differentially Expressed Genes
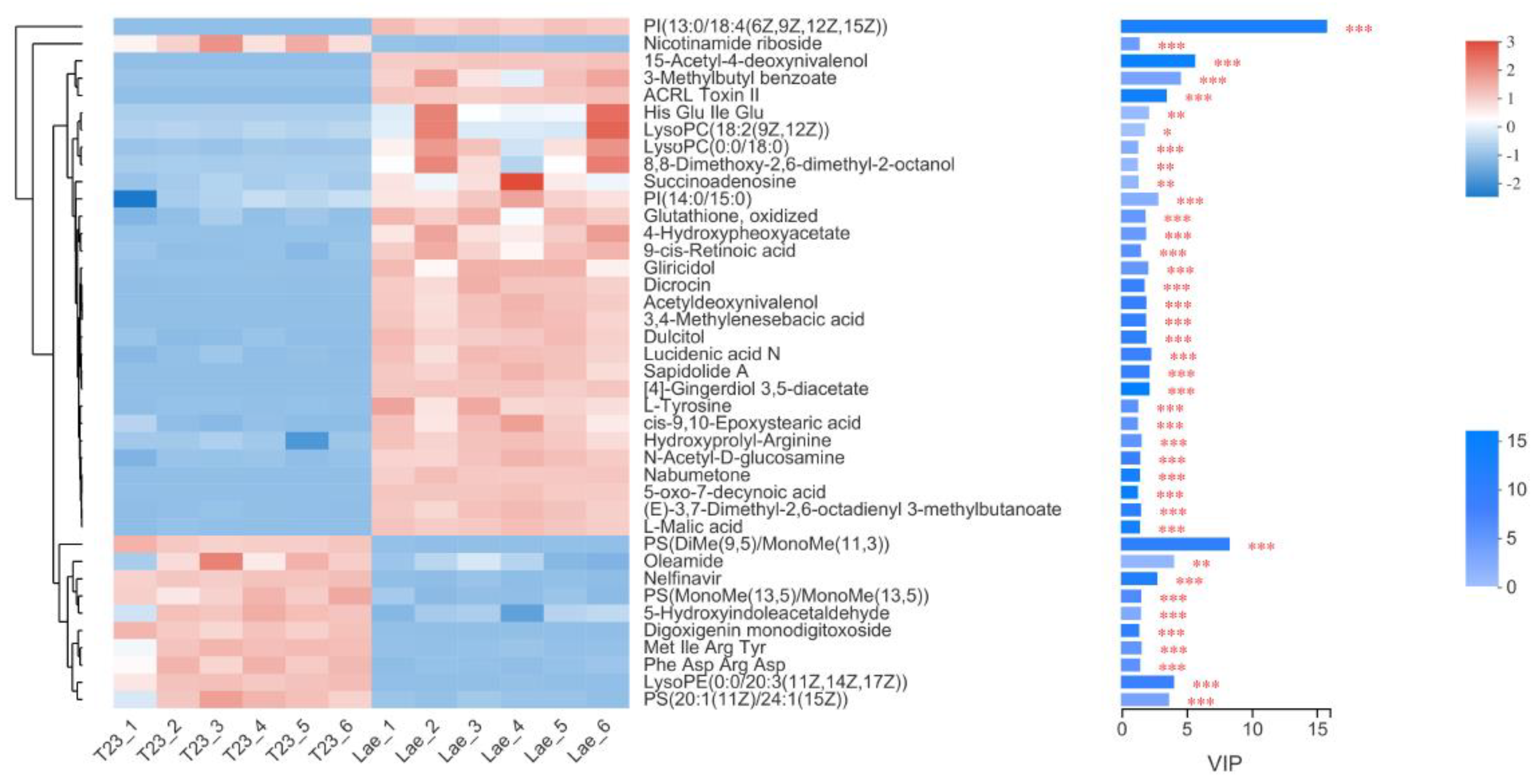
3.4. Metabolome Difference Analysis
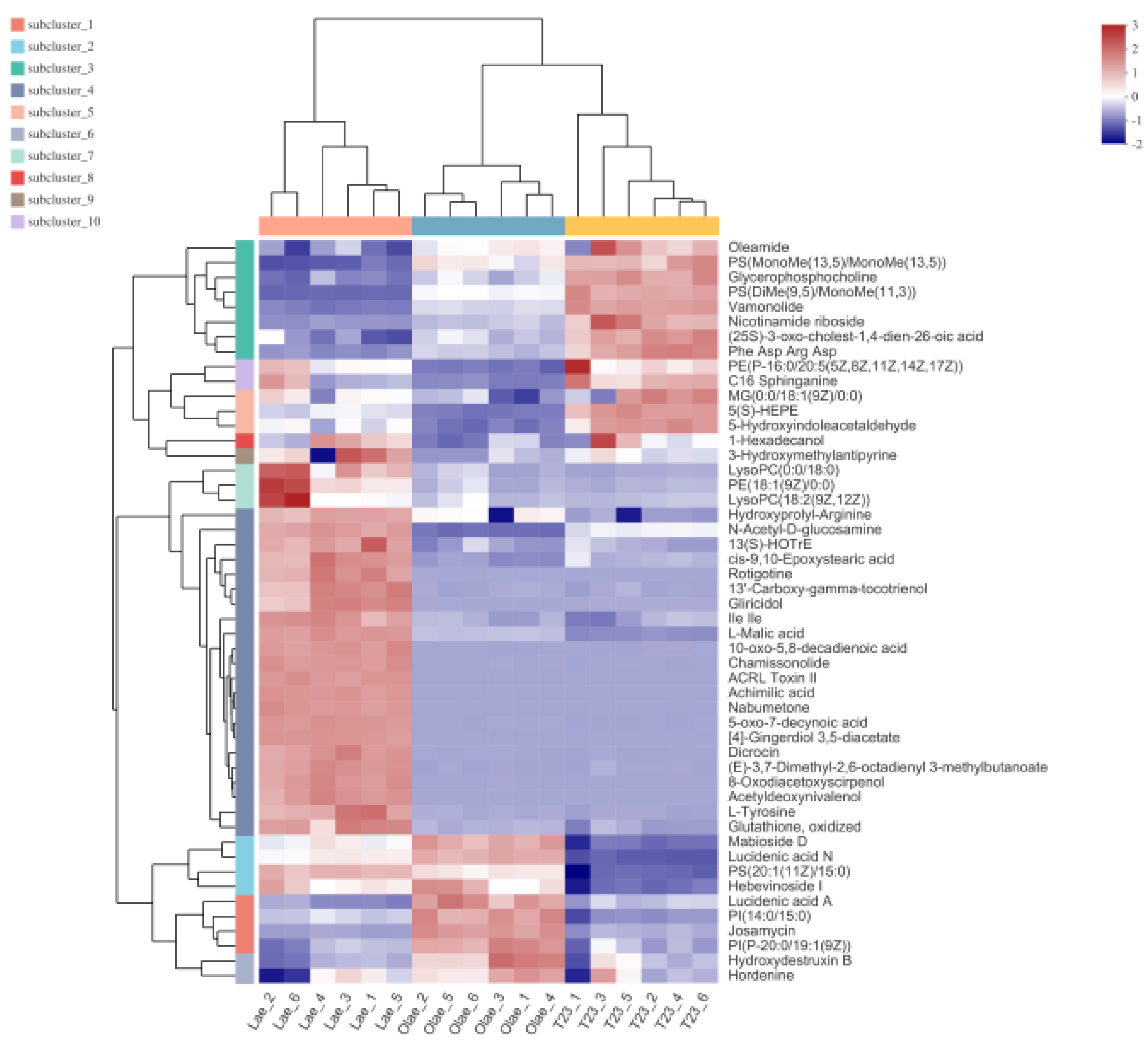
3.5. Analysis of Differential Metabolites
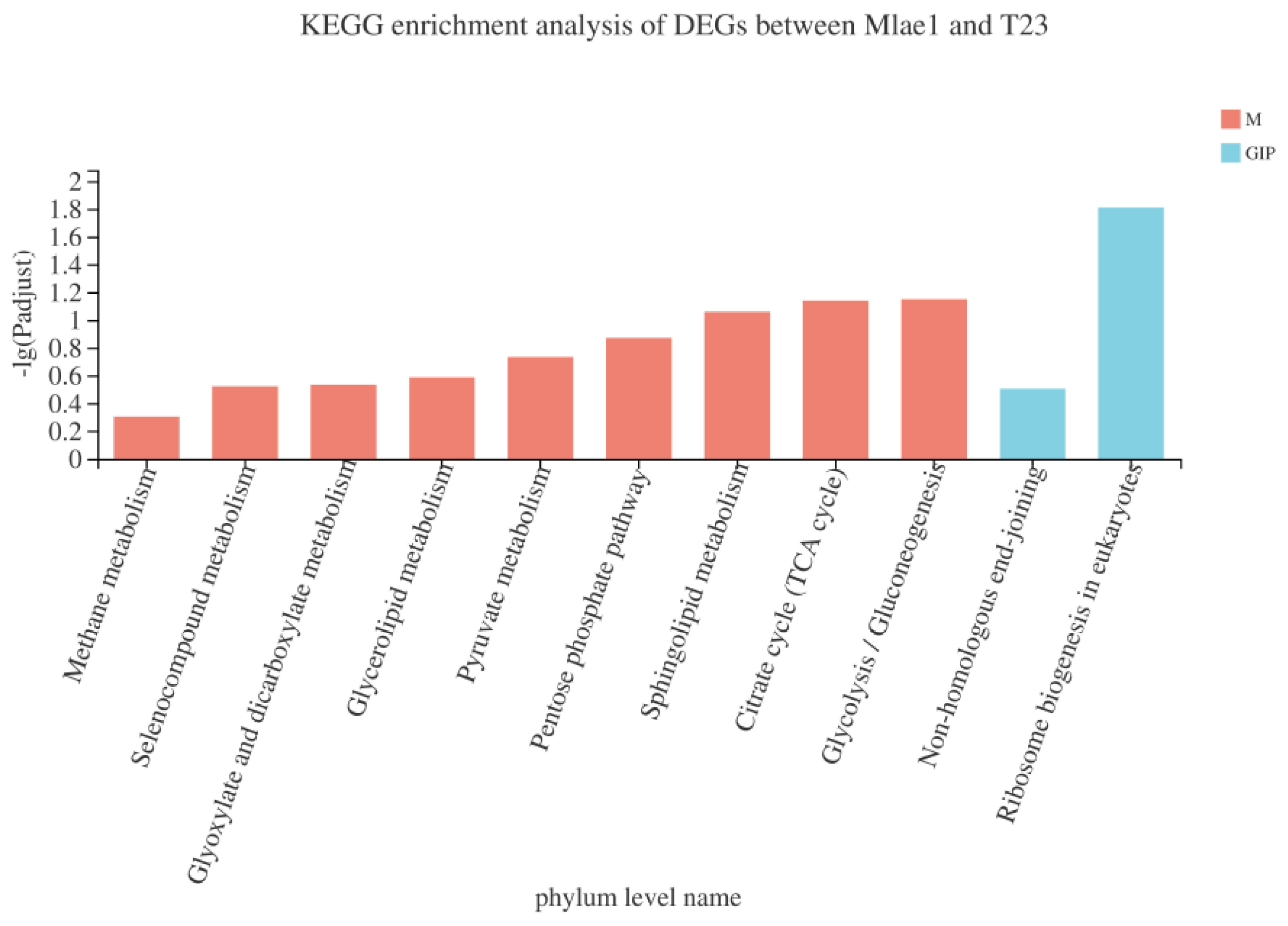
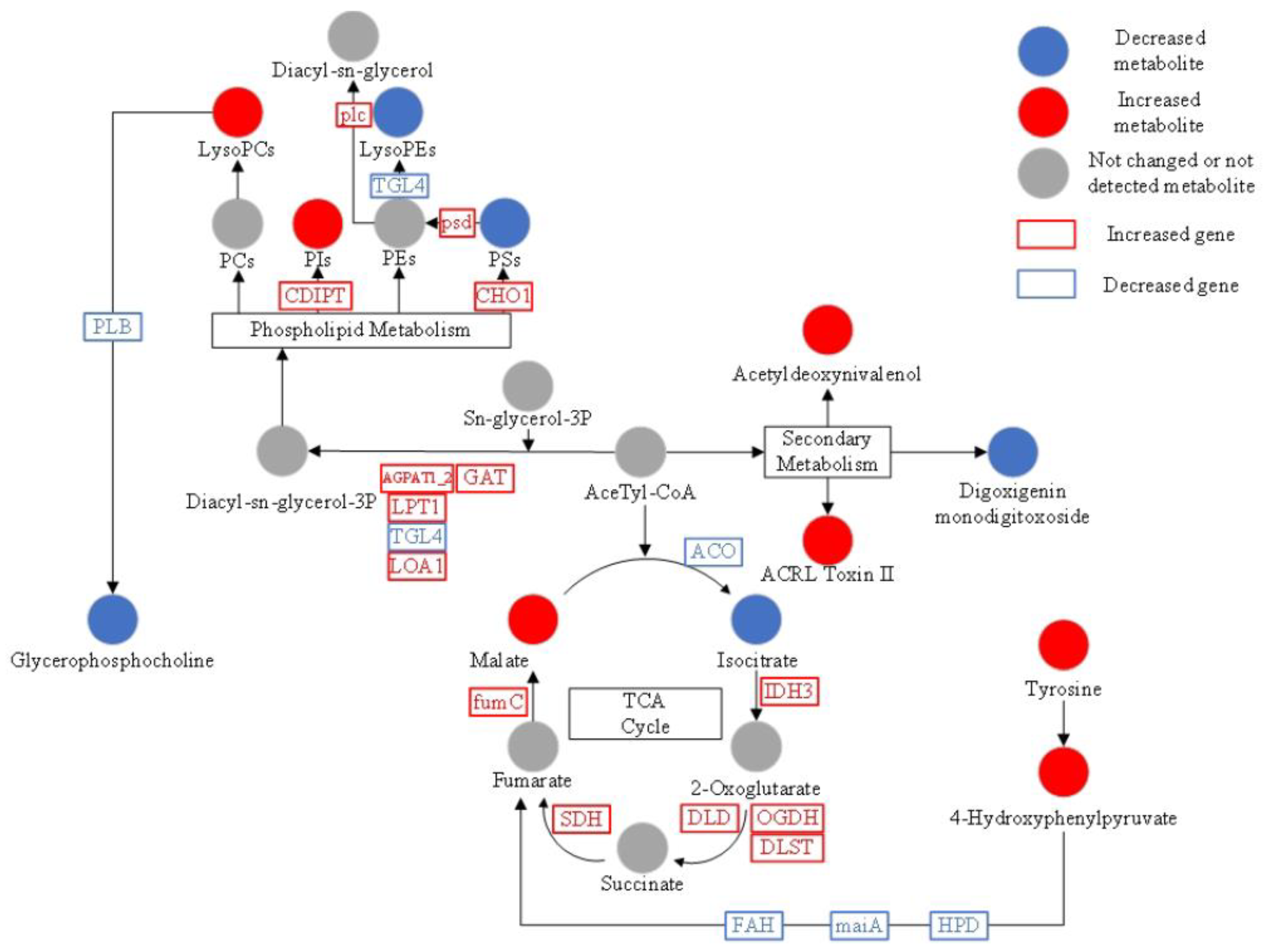
3.6. KEGG Analysis of Differential Metabolites
4. Discussion
4.1. Integrated Analysis of Metabolic Pathways
4.2. Lae1 Regulates Phospholipid Metabolism
4.3. Lae1 Gene Regulates Phosphatidylinositol Signaling System
4.4. Changes in Phospholipid Metabolism Affects Membrane Composition
4.5. ATP Synthase Genes Were Upregulated in Mlae1
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, W.Y.; Liu, Y.; Lin, Y.T.; Guo, K.; Li, X.N.; Luo, S.H.; Li, S.H. Antibacterial harziane diterpenoids from a fungal symbiont Trichoderma atroviride isolated from Colquhounia coccinea var. mollis. Phytochemistry 2020, 170, 112198. [Google Scholar] [CrossRef]
- Lorito, M.; Woo, S.L.; Harman, G.E.; Monte, E. Translational Research on Trichoderma: From ‘Omics to the Field. Annu. Rev. Phytopathol. 2010, 48, 395–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.L.; Ruocco, M.; Vinale, F.; Nigro, M.; Marra, R.; Lombardi, N.; Pascale, A.; Lanzuise, S.; Manganiello, G.; Lorito, M. Trichoderma-based Products and their Widespread Use in Agriculture. Open Mycol. J. 2014, 8, 71–126. [Google Scholar] [CrossRef] [Green Version]
- Graczyk, S.; Perlińska-Lenart, U.; Górka-Nieć, W.; Lichota, R.; Piłsyk, S.; Zembek, P.; Lenart, J.; Bernat, P.; Gryz, E.; Augustyniak, J.; et al. Increased activity of the sterol branch of the mevalonate pathway elevates glycosylation of secretory proteins and improves antifungal properties of Trichoderma atroviride. Fungal Genet. Biol. 2020, 137, 103334. [Google Scholar] [CrossRef]
- Mukherjee, M.; Mukherjee, P.K.; Horwitz, B.A.; Zachow, C.; Berg, G.; Zeilinger, S. Trichoderma–Plant–Pathogen Interactions: Advances in Genetics of Biological Control. Indian J. Microbiol. 2012, 52, 522–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayram, O.; Krappmann, S.; Ni, M.; Bok, J.W.; Helmstaedt, K.; Valerius, O.; Braus-Stromeyer, S.; Kwon, N.-J.; Keller, N.P.; Yu, J.-H.; et al. VelB/VeA/LaeA Complex Coordinates Light Signal with Fungal Development and Secondary Metabolism. Science 2008, 320, 1504–1506. [Google Scholar] [CrossRef]
- Jain, S.; Keller, N. Insights to fungal biology through LaeA sleuthing. Fungal Biol. Rev. 2013, 27, 51–59. [Google Scholar] [CrossRef]
- Bok, J.W.; Chiang, Y.M.; Szewczyk, E.; Reyes-Dominguez, Y.; Davidson, A.D.; Sanchez, J.F.; Lo, H.-C.; Watanabe, K.; Strauss, J.; Oakley, B. Chromatin-level regulation of biosynthetic gene clusters. Nat. Chem. Biol. 2009, 5, 462–464. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.-H.; Keller, N. Regulation of Secondary Metabolism in Filamentous Fungi. Annu. Rev. Phytopathol. 2005, 43, 437–458. [Google Scholar] [CrossRef] [PubMed]
- Bok, J.W.; Hoffmeister, D.; Maggio-Hall, L.A.; Murillo, R.; Glasner, J.D.; Keller, N.P. Genomic Mining for Aspergillus Natural Products. Chem. Biol. 2006, 13, 31–37. [Google Scholar] [CrossRef]
- Perrin, R.M.; Fedorova, N.D.; Bok, J.W.; Cramer, R.A., Jr.; Wortman, J.R.; Kim, H.S.; Nierman, W.C.; Keller, N.P. Transcriptional Regulation of Chemical Diversity in Aspergillus fumigatus by LaeA. PLoS Pathog. 2007, 3, e50. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Lv, Y.; Li, X.; Lin, Y.; Deng, H.; Pan, L. Profiling of secondary metabolite gene clusters regulated by LaeA in Aspergillus niger FGSC A1279 based on genome sequencing and transcriptome analysis. Res. Microbiol. 2018, 169, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-K.; Scharfenstein, L.L.; Ehrlich, K.C.; Wei, Q.; Bhatnagar, D.; Ingber, B.F. Effects of laeA deletion on Aspergillus flavus conidial development and hydrophobicity may contribute to loss of aflatoxin production. Fungal Biol. 2012, 116, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Yun, Y.; Liu, Y.; Ma, Z. FgVELB is associated with vegetative differentiation, secondary metabolism and virulence in Fusarium graminearum. Fungal Genet. Biol. 2012, 49, 653–662. [Google Scholar] [CrossRef]
- Wiemann, P.; Brown, D.W.; Kleigrewe, K.; Bok, J.W.; Keller, N.P.; Humpf, H.-U.; Tudzynski, B. FfVel1 and FfLae1, components of a velvet-like complex in Fusarium fujikuroi, affect differentiation, secondary metabolism and virulence. Mol. Microbiol. 2010, 77, 972–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Oide, S.; Zhang, N.; Choi, M.Y.; Turgeon, B.G. ChLae1 and ChVel1 Regulate T-toxin Production, Virulence, Oxidative Stress Response, and Development of the Maize Pathogen Cochliobolus heterostrophus. PLOS Pathog. 2012, 8, e1002542. [Google Scholar] [CrossRef] [Green Version]
- Seiboth, B.; Karimi, R.A.; Phatale, P.A.; Linke, R.; Hartl, L.; Sauer, D.G.; Smith, K.M.; Baker, S.E.; Freitag, M.; Kubicek, C.P. The putative protein methyltransferase LAE1 controls cellulase gene expression in Trichoderma reesei. Mol. Microbiol. 2012, 84, 1150–1164. [Google Scholar] [CrossRef]
- Reyes-Dominguez, Y.; Bok, J.W.; Berger, H.; Shwab, E.K.; Basheer, A.; Gallmetzer, A.; Scazzocchio, C.; Keller, N.; Strauss, J. Heterochromatic marks are associated with the repression of secondary metabolism clusters in Aspergillus nidulans. Mol. Microbiol. 2010, 76, 1376–1386. [Google Scholar] [CrossRef] [Green Version]
- Karimi-Aghcheh, R.; Bok, J.W.; Phatale, P.A.; Smith, K.M.; Baker, S.E.; Lichius, A.; Omann, M.; Zeilinger, S.; Seiboth, B.; Rhee, C.; et al. Functional Analyses of Trichoderma reesei LAE1 Reveal Conserved and Contrasting Roles of This Regulator. G3 Genes Genomes Genet. 2013, 3, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Butchko, R.A.; Brown, D.W.; Busman, M.; Tudzynski, B.; Wiemann, P. Lae1 regulates expression of multiple secondary metabolite gene clusters in Fusarium verticillioides. Fungal Genet. Biol. 2012, 49, 602–612. [Google Scholar] [CrossRef]
- Fekete, E.; Karaffa, L.; Karimi Aghcheh, R.; Németh, Z.; Fekete, É.; Orosz, A.; Paholcsek, M.; Stágel, A.; Kubicek, C.P. The transcriptome of lae1 mutants of Trichoderma reesei cultivated at constant growth rates reveals new targets of LAE1 function. BMC Genom. 2014, 15, 447. [Google Scholar] [CrossRef] [Green Version]
- Niehaus, E.M.; Rindermann, L.; Janevska, S.; Münsterkötter, M.; Güldener, U.; Tudzynski, B. Analysis of the global regulator Lae1 uncovers a connection between Lae1 and the histone acetyltransferase HAT1 in Fusarium fujikuroi. Appl. Microbiol. Biotechnol. 2018, 102, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.-C.; Shi, W.-L.; Zhou, Y.-R.; Chen, X.-L.; Zhang, Y.-Z.; Zhang, X.; Zhang, W.-X.; Song, X.-Y. The Putative Methyltransferase TlLAE1 Is Involved in the Regulation of Peptaibols Production in the Biocontrol Fungus Trichoderma longibrachiatum SMF2. Front. Microbiol. 2020, 11, 1267. [Google Scholar] [CrossRef]
- Motoyama, T.; Ishii, T.; Kamakura, T.; Osada, H. Screening of tenuazonic acid production-inducing compounds and identification of NPD938 as a regulator of fungal secondary metabolism. Biosci. Biotechnol. Biochem. 2021, 85, 2200–2208. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Chen, X.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar]
- Jarsch, I.K.; Daste, F.; Gallop, J.L. Membrane curvature in cell biology: An integration of molecular mechanisms. J. Cell Biol. 2016, 214, 375–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardsley-Elliot, A.; Plosker, G.L. Nelfinavir. Drugs 2000, 59, 581–620. [Google Scholar] [CrossRef] [PubMed]
- Hedner, T.; Samulesson, O.; Währborg, P.; Wadenvik, H.; Ung, K.A.; Ekbom, A. Nabumetone. Drugs 2004, 64, 2315–2343. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, N.A.; Wellington, K.; Easthope, S.E. Rotigotine. CNS Drugs 2005, 19, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Bordoloi, M.; Barua, N.C.; Mohan, S.; Dutta, S.C.; Mathur, R.K.; Ghosh, A.C.; Rychlewska, U. Sapidolide A: An unprecedented spherical carbocyclic lactone from Baccaurea sapida seed kernels: Is it a meroisoprenoid? Tetrahedron Lett. 1996, 37, 6791–6792. [Google Scholar] [CrossRef]
- Drake, T.A.; Ping, P. Thematic review series: Systems Biology Approaches to Metabolic and Cardiovascular Disorders.Proteomics approaches to the systems biology of cardiovascular diseases. J. Lipid Res. 2006, 48, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mironenka, J.; Różalska, S.; Soboń, A.; Bernat, P. Lipids, proteins and extracellular metabolites of Trichoderma harzianum modifications caused by 2,4-dichlorophenoxyacetic acid as a plant growth stimulator. Ecotoxicol. Environ. Saf. 2020, 194, 110383. [Google Scholar] [CrossRef]
- Gillaspy, G.E. The cellular language of myo-inositol signaling. New Phytol. 2011, 192, 823–839. [Google Scholar] [CrossRef]
- Huo, J.-F.; Song, S.-S.; Li, X.; Yang, W.-X.; Fu, S.-J. Preliminary Research on Ca2+··CaM Involved in the Resistance Response of Wheat to Puccinia triticina. Acta Agric. Boreali-Sin. 2008, 23, 234–239. [Google Scholar]
- Li, Z.; Agellon, L.B.; Allen, T.M.; Umeda, M.; Jewell, L.; Mason, A.; Vance, D.E. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006, 3, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Nykiel-Szymańska, J.; Różalska, S.; Bernat, P.; Słaba, M. Assessment of oxidative stress and phospholipids alterations in chloroacetanilides-degrading Trichoderma spp. Ecotoxicol. Environ. Saf. 2019, 184, 109629. [Google Scholar] [CrossRef] [PubMed]
- Kapri-Pardes, E.; Katz, A.; Haviv, H.; Mahmmoud, Y.; Ilan, M.; Khalfin-Penigel, I.; Carmeli, S.; Yarden, O.; Karlish, S.J.D. Stabilization of the α2 Isoform of Na,K-ATPase by Mutations in a Phospholipid Binding Pocket. J. Biol. Chem. 2011, 286, 42888–42899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, D.; Vicôgne, J.; Zaitseva, I.; Zaitseva, S.; Pessin, J.E. Evidence that electrostatic interactions between vesicle-associated membrane protein 2 and acidic phospholipids may modulate the fusion of transport vesicles with the plasma membrane. Mol. Biol. Cell 2009, 20, 4910–4919. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Campbell, H.A.; King, S.C.; Dowhan, W. Phospholipids as determinants of membrane protein topology: Phosphatidylethanolamine is required for the proper topological organization of the γ-aminobutyric acid permease (GabP) of Escherichia coli. J. Biol. Chem. 2005, 280, 26032–26038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghcheh, R.K.; Druzhinina, I.S.; Kubicek, C.P. The Putative Protein Methyltransferase LAE1 of Trichoderma atroviride Is a Key Regulator of Asexual Development and Mycoparasitism. PLoS ONE 2013, 8, e67144. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Formula | VIP | FC (T23/Mlae1) | p-Value |
|---|---|---|---|---|
| 15-Acetyl-4-deoxynivalenol | C17H22O7 | 5.6182 | 0.0059 | 1.83 × 10−15 |
| ACRL Toxin II | C17H24O5 | 3.4046 | 0.0013 | 1.43 × 10−14 |
| Nelfinavir | C32H45N3O4S | 2.6966 | 2.6108 | 5.47 × 10−13 |
| Sapidolide A | C14H18O5 | 2.089 | 0.0001 | 1.91 × 10−10 |
| Gliricidol | C16H16O7 | 1.9971 | 0.0135 | 1.21 × 10−6 |
| Acetyldeoxynivalenol | C17H22O7 | 1.8362 | 0 | 2.28 × 10−10 |
| Nabumetone | C15H16O2 | 1.347 | 0.0005 | 4.98 × 10−14 |
| Digoxigenin monodigitoxoside | C29H44O8 | 1.2926 | 4.6022 | 3.73 × 10−11 |
| cis-9,10-Epoxystearic acid | C18H34O | 1.1944 | 0.4748 | 5.80 × 10−7 |
| Rotigotine | C19H25NOS | 1.0502 | 0.0006 | 3.85 × 10−8 |
| Metabolite | Formula | VIP | FC (T23/Olae1) | p-Value |
|---|---|---|---|---|
| Josamycin | C42H69NO15 | 2.5971 | 0.244 | 9.84 × 10−11 |
| Maduramicin | C47H80O17 | 1.0745 | 0.2895 | 1.25 × 10−10 |
| Lucidenic acid A | C27H38O6 | 2.0008 | 0.5255 | 1.00 × 10−6 |
| Hydroxydestruxin B | C30H51N5O8 | 1.1761 | 0.6212 | 1.66 × 10−3 |
| Ergothioneine | C9H15N3O2S | 1.1284 | 1.387 | 2.15 × 10−5 |
| Isocitrate | C6H8O7 | 2.1805 | 1.4663 | 8.75 × 10−11 |
| Pentamidine | C19H24N4O2 | 1.1724 | 1.8769 | 4.26 × 10−8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Y.; Zhang, Y.; Zhang, H.; Wang, X.; Chen, J.; Li, Y. Integrated Transcriptome and Untargeted Metabolomic Analyses Revealed the Role of Methyltransferase Lae1 in the Regulation of Phospholipid Metabolism in Trichoderma atroviride. J. Fungi 2023, 9, 120. https://doi.org/10.3390/jof9010120
Shen Y, Zhang Y, Zhang H, Wang X, Chen J, Li Y. Integrated Transcriptome and Untargeted Metabolomic Analyses Revealed the Role of Methyltransferase Lae1 in the Regulation of Phospholipid Metabolism in Trichoderma atroviride. Journal of Fungi. 2023; 9(1):120. https://doi.org/10.3390/jof9010120
Chicago/Turabian StyleShen, Yanxiang, Yiwen Zhang, Hui Zhang, Xinhua Wang, Jie Chen, and Yaqian Li. 2023. "Integrated Transcriptome and Untargeted Metabolomic Analyses Revealed the Role of Methyltransferase Lae1 in the Regulation of Phospholipid Metabolism in Trichoderma atroviride" Journal of Fungi 9, no. 1: 120. https://doi.org/10.3390/jof9010120