1. Introduction
The yeast vacuole is a hub for degradation and signal transduction of many pathways [
1]. Endocytosis and macroautophagy are typical pathways to deliver cargos to vacuoles in yeast and to lysosomes in mammalian cells [
2]. Endocytosis is a process to actively internalize molecules and surface proteins via an endocytic vesicle, which may end up in lysosomes/vacuoles [
3]. Macroautophagy is a process to engulf cargoes in double-membrane phagophores (precursors of autophagosomes or unclosed autophagosomes), which are sealed to form autophagosomes and to fuse with lysosomes/vacuoles to degrade the enclosed materials for recycling purposes [
4]. In contrast to macroautophagy, eukaryotic cells directly degrade a wide range of autophagic cargoes through microautophagy, which has emerging mechanisms and functions and has drawn much attention to the autophagy field [
5,
6]. Both endocytosis and macroautophagy (hereafter referred to as autophagy) are associated with physiopathology and cell survival under different stress conditions. Both vesicles from endocytosis and autophagosomes from autophagy can fuse with lysosomes/vacuoles. These fusion steps are positively regulated by the fusion machinery, including but not limited to Rab7/Ypt7, homotypic fusion and vacuole protein sorting (HOPS) tethering complexes and soluble N-ethylmaleimide sensitive fusion protein (NSF) attachment receptors (SNAREs) [
7,
8,
9]. The absence of any of these regulators inhibits the fusion process.
The yeast endosomal Rab5 GTPase Vps21 [
10] and the endosomal sorting complex required for transport (ESCRT), which is composed of conserved multiple protein complexes, ESCRT-0, ESCRT-I, ESCRT-II, ESCRT-III, and a number of accessory proteins (e.g., the Vps4 ATPase) [
11], play roles before the fusion step with vacuoles in both endocytosis and autophagy pathways [
9,
12]. The absence of Vps21 and ESCRT subunits results in phagophore accumulation under a short duration of autophagy induction [
12,
13,
14]. However, these phagophores entered vacuoles under a prolonged duration of nitrogen starvation [
15]. Previously, only closed autophagosomes were supposed to enter vacuoles/lysosomes [
16]. However, recently, it became clear that phagophores can also fuse with lysosomes or vacuoles in mutant yeast and mammalian cells, although the molecular machinery is unknown [
4,
15,
17]. Failure of autophagosome biogenesis and phagophore closure results in an interrupted autophagy process, which compromises homeostasis and leads to various diseases, including metabolic disorders, neurodegeneration and cancer [
18]. The fusion of phagophores with lysosomes/vacuoles partially restores cargo degradation and recycling, maintaining autophagy to a certain degree to resist stresses [
4,
15]. In response to nitrogen starvation in the presence of a poor carbon source, diploid cells of
S. cerevisiae undergo meiosis and package the haploid nuclei produced in meiosis into a tetrad with four spores [
19]. The
vps21∆/
vps21∆ diploid cells are with phagophore accumulation of autophagy defect during a short duration but with near-complete autophagy and sporulation ability during a long duration of nitrogen starvation, which are different from the loss of sporulation ability in autophagy completely impaired mutant (
atg1∆/
atg1∆ and
pep4∆/
pep4∆) diploid cells, providing solid evidence for the physiological roles of phagophore–vacuole fusion [
15].
The vacuolar transport chaperon (VTC) complex is a novel family of yeast chaperons (including Vtc1-5) mainly involved in the synthesis and transfer of polyP to vacuole, the distribution of V-ATPase and other membrane proteins, microautophagy, and membrane trafficking [
20,
21,
22,
23]. VTC complex proteins are negative regulators for the entry of misfolded glycosylphosphatidylinositol (GPI)-anchored proteins into vacuoles with microautophagy [
24]. We recently found that in
vps21∆ cells, the depletion of most Vtc proteins (four of five) promoted the entry of phagophores into vacuoles, while the depletion of Ypt7 and Vam3 in
vps21∆ cells inhibited this process, by determining the changes in GFP-Atg8 vacuolar localization and degradation [
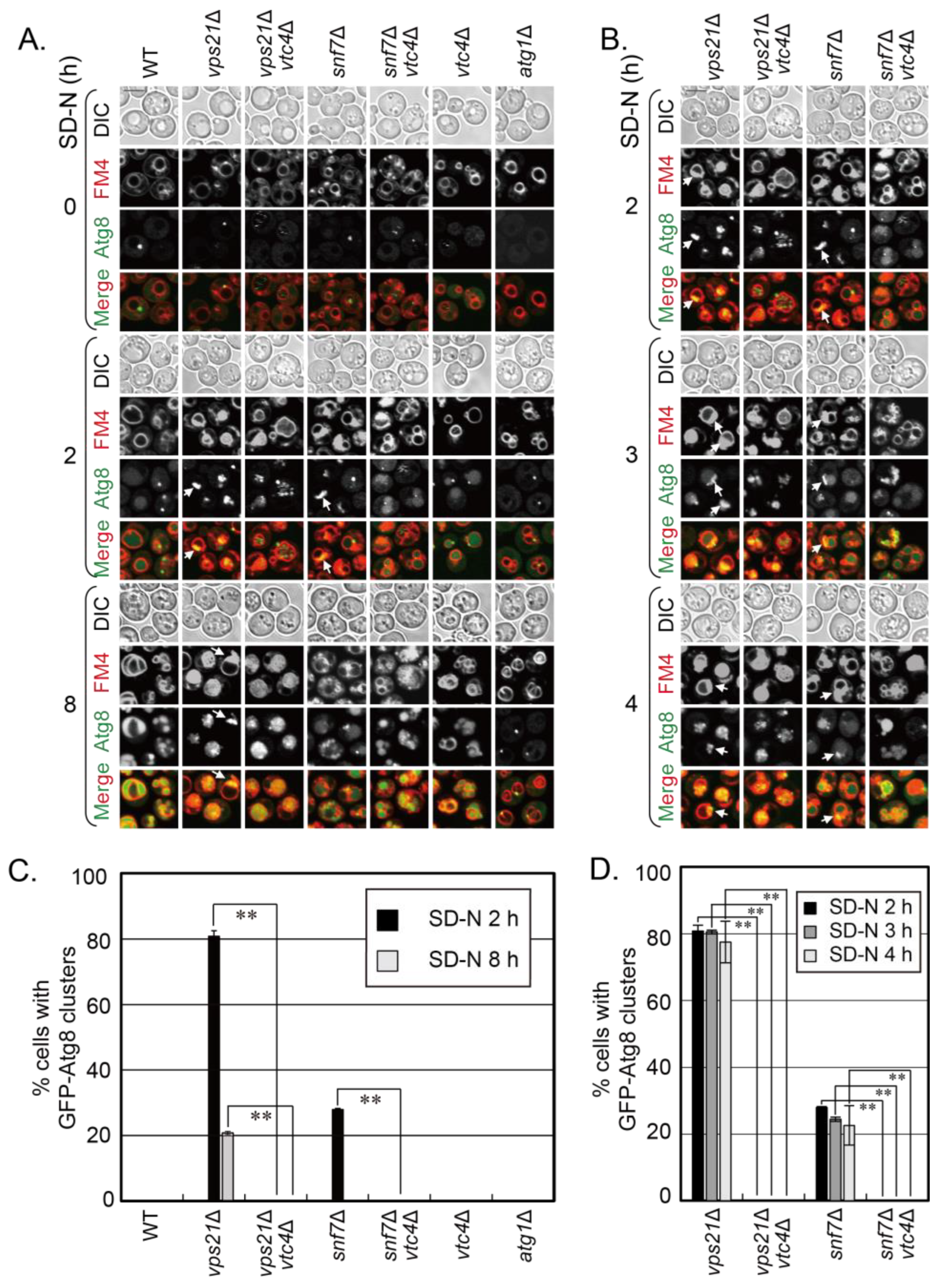
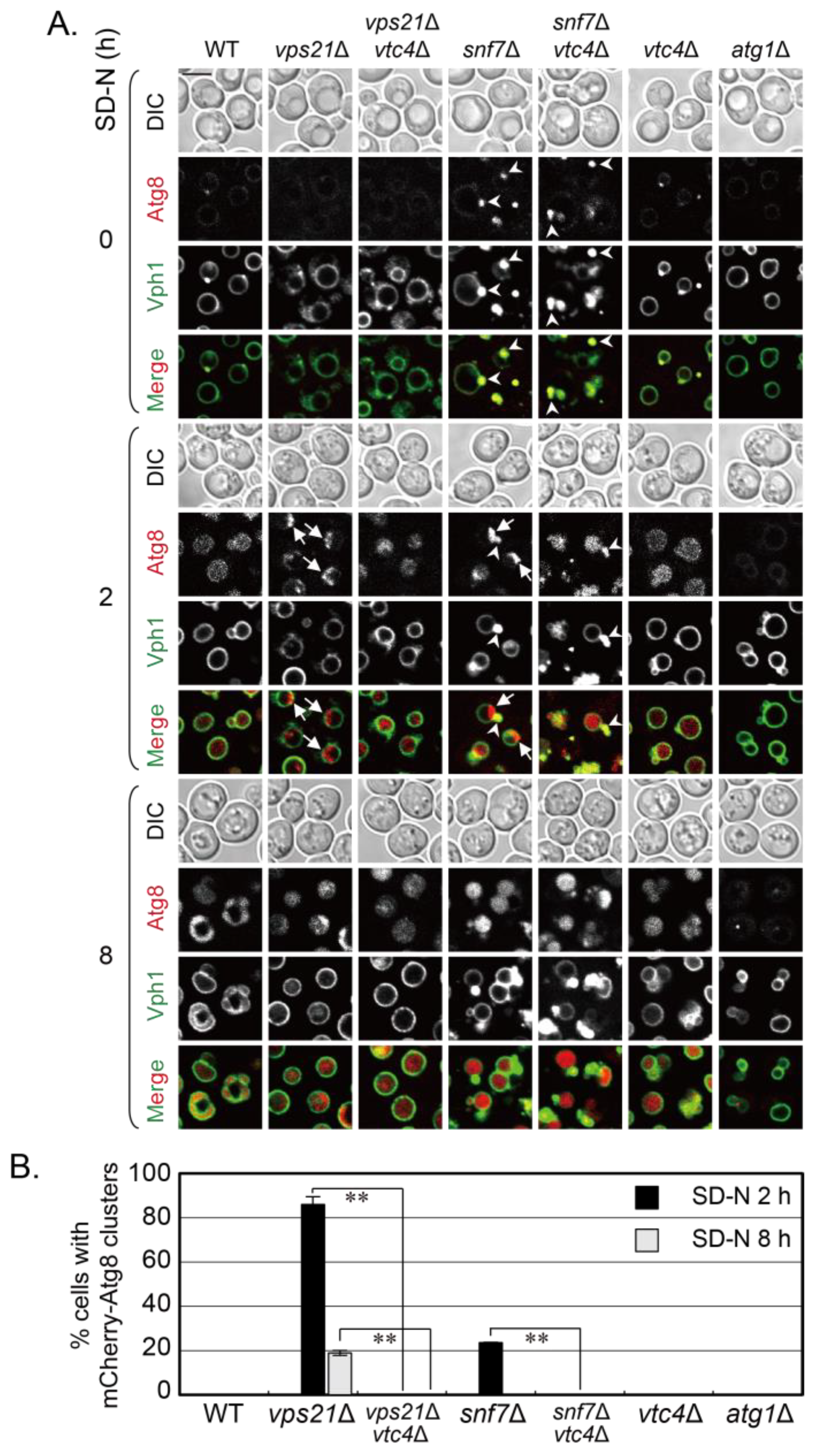
15]. However, it is unknown whether VTC complex proteins have a general negative regulatory role in promoting proteins, complexes, vesicles, or bigger membrane structures into vacuoles. We reported that the absence of Vps21 and ESCRT subunits in yeast cells resulted in similar phagophore accumulation during autophagy [
12]. We were curious to know whether the depletion of VTC complex proteins also promoted the entry of phagophores in ESCRT mutant cells into vacuoles. Snf7 is a core subunit of ESCRT-III, and phagophores accumulate in
snf7∆ cells under a short duration of nitrogen starvation [
12]. We removed a representative VTC complex protein Vtc4 from
snf7∆ cells as from
vps21∆ cells and similarly examined phenotypes [
15]. We examined the vacuolar localization and degradation of phagophores in
snf7∆ and
snf7∆
vtc4∆ cells, in which phagophores were shown with green fluorescence protein (GFP) labeled Atg8, and vacuoles were stained with lipophilic dye FM4-64 or labeled with Vph1-GFP. We used
vps21∆ and
vps21∆
vtc4∆ cells as controls to show the promotion of depletion of VTC complex proteins in phagophore entry into vacuoles. We found that Vtc4 depletion also promoted the entry of phagophores in
snf7∆ cells into vacuoles as it did for
vps21∆ cells, although it is more obvious in
vps21∆ cells. These results revalidate the role of VTC complex proteins in negatively regulating phagophore entry into vacuoles and may also indicate a widespread role of the VTC complex in negatively regulating the entry of other cargos into yeast vacuoles.
2. Materials and Methods
2.1. Strains and Reagents
The
Saccharomyces cerevisiae yeast strains used in this study are listed in
Table S1 [
9,
15,
25]. All yeast and
Escherichia coli transformations were performed as previously described [
26].
The parent
S. cerevisiae yeast strain SEY6210 was used for fluorescence tagging and gene deletion to generate strains for this study. GFP-Atg8 expressed from the
pP1KGreen fluorescent protein (GFP)-ATG8(406) was constructed with 990 base pairs of
ATG8 5′ sequence in front of the GFP-Atg8 open reading frame and linearized with EcoRV and integrated into the target strains [
27]. GFP-Atg8- or mCherry-Atg8-tagged strains were confirmed for fluorescence phenotypes, and Vph1-GFP strains were further tagged with a
HIS3 cassette for mCherry-Atg8-tagged strains. A drug-resistance cassette (
hphMX4 or
kanMX3) was used to replace the targeted gene in different background strains to generate deletion strains. Either Vph1-GFP tagging or gene deletion was achieved by using polymerase chain reaction (PCR) amplification with corresponding primers in
Table S2 to produce DNA fragments, in combination with DNA fragment transformation for endogenous recombination. Vph1-GFP-tagged strains were selected on SD-His plates, and gene deletion strains were selected on YPD+Hygromycin B (H8080, Solarbio, Beijing, China) or YPD+Geneticin (G8160, Solarbio), depending on the drug-resistance cassette
hphMX4 or
kanMX3, respectively. The deletion strains were further confirmed with diagnostic PCR with corresponding primers in
Table S2. The function of Vtc4 depletion in promoting the entry of phagophores into vacuoles in Vps21 or Snf7 depletion strain was examined with fluorescence microscopy observations and immunoblotting assays after the cells were grown under normal growth conditions or induced for autophagy.
Antibodies used in this study were mouse anti-GFP (sc-9996; Santa Cruz Biotechnology, Dallas, TX, USA), rabbit anti-glucose-6-phosphate dehydrogenase (G6PDH, A9521; Sigma-Aldrich, St. Louis, MO, USA), horseradish peroxidase (HRP)-linked goat anti-mouse Immunoglobulin G (IgG) (GAM007; Multisciences, Hangzhou, China), and HRP-linked goat anti-rabbit IgG (GAR007; Multisciences). All other chemical reagents used in this study were purchased from Sigma (St Louis, MO, USA) or from Amersco (Fair Lawn, NJ, USA), except for SynaptoRed, also known as FM4-64 from Biotium (70021; Hayward, CA, USA), and PCR polymerases and buffers from Takara Biotechnology (Dalian, China).
2.2. Yeast Growth, Induction of Autophagy and Microscopy Observation
Strains were grown on rich yeast extract peptone dextrose (YPD) plates or in YPD liquid at 26 °C for 3 days. If the strains were deletion strains, an additional 0.02% Geneticin or 0.03% Hygromycin B was added into medium. YPD liquid contains 2% (w/v) peptone, 1% yeast extract, 2% glucose, with an additional 2% agar for plate. The overnight culture was consecutively re-inoculated for three times at an initial optical density at 600 nm (OD600) of approximately 0.005–0.015 in 3 mL of YPD medium with rotation at 200 rpm to reach the mid-log phase for live-cell fluorescence microscopy.
Autophagy was induced by incubating cells in a synthetic liquid medium without nitrogen (SD-N). This medium comprised 0.17% yeast nitrogen base without amino acids and ammonium sulfate and 2% glucose. Briefly, cells were collected at the mid-log phase by centrifugation at 4000 rpm for 3 min. The supernatant was removed to add an equal volume of sterilized water to resuspend and wash away the nutrient from the pelleted cells for three repeats. The final supernatant was removed after centrifugation and replaced with a 60% volume of SD-N medium for starvation. The new culture was incubated for the indicated periods to observe fluorescence images and/or perform immunoblotting assays.
A final concentration of 1.6 μM FM4-64 was added in the indicated culture, and the culture was incubated at 26 °C for 2 h to stain the cell vacuoles before collecting the cells. The cells were examined on slides using a Leica (Wetzlar, Germany) laser scanning confocal microscope (TCS SP8), equipped with a high-contrast Plan Apochromat 100 × 1.4-NA oil-immersion objective lens, photomultiplier tube detectors and Hybrid detectors, and acquisition software Leica LAS X. More than three fields per sample were assessed. Those fluorescence images presented along with differential interference contrast (DIC) images were acquired using a Leica confocal microscope with LAS X program for single slices, and a central slice was selected. Images were processed for brightness and contrast with Photoshop CS6 (Adobe, San Jose, CA, USA) and assembled with Illustrator CS5 (Adobe) for publication [
12].
2.3. Immunoblotting Analysis
Crude lysates from cells expressing GFP-Atg8 cells were subjected to immunoblotting assay to analyze autophagy processing in at least two independent experiments, as previously described [
28,
29]. Blots were probed with an anti-GFP antibody to determine the levels of GFP-Atg8 and GFP. An anti-G6PDH antibody was used to determine the expression levels of glucose-6-phosphate dehydrogenase (G6PDH) to display the loading situation. Immunoblots performed with indicated antibodies were subjected to electrochemiluminescence (ECL) HRP substrate (P90720; Millipore, Burlington, MA, USA) and exposed to X-ray film (XBT; Carestream, Rochester, NY, USA) to generate an image with an HQ-350XT film developer (Huqiu Imaging Technology, Suzhou, China). Immunoblotting bands were quantified using the ImageJ (National Institutes of Health, Bethesda, MD, USA) for band density in cases where the quantification was necessary. The relative percentage of GFP accounted for the total amount of GFP-Atg8, and GFP [(% GFP = (band density of GFP)/(band density of GFP-Atg8 + band density of GFP) × 100%] was used to indicate the autophagy level. Therefore, there is no need to normalize GFP-Atg8 and GFP bands to G6PDH loading band for each sample.
2.4. Statistical Analyses
The data are reported as the mean ± standard deviation (SD) calculated from at least two independent experiments. Student’s t-test was used to determine statistical significance. The p-value was used to demonstrate the significance, which was represented as: n.s., not significant; and **, p < 0.01.
4. Discussion
The entry of cargoes into vacuoles is regulated by multiple pathways and regulators in yeast cells. The fusion step is essential for cargoes in membrane structures to enter vacuoles. There are many positive regulators for vesicle–vacuole fusions in the endocytosis pathway or autophagosome–vacuole fusions in autophagy, such as a yeast Ypt7 module, including GEF subunits, Mon1 and Ccz1; Rab GTPase, Ypt7; tether, HOPS complex; SNARE protein, Vam3 [
9]. Previously, only closed autophagosomes were supposed to fuse with vacuoles/lysosomes to be degraded [
16]. In two-dimensional topology thinking, the double-membrane phagophores were thought to be unable to fuse with lysosomes/vacuoles to degrade enclosed cargoes. After the fusion of the outer membrane of the phagophore with the vacuolar membrane, it was thought that the cargoes inside phagophores would be expelled out. However, the autophagosome–vacuole fusion is a three-dimensional process, and the supposed expelling process will not occur autonomously [
4]. Recently, phagophores were found to fuse with lysosomes in mammalian cells or with vacuoles in yeast cells [
4,
15,
17]. The phagophores in vacuoles in
vps21∆ and ESCRT mutant cells clearly indicate that phagophores enter vacuoles [
15]. The detailed mechanisms for these fusion processes are still unknown; however, these three-dimensional fusion processes should not be denied with incorrect two-dimensional topology thinking [
4]. Ypt7 and Vam3 are required for the fusion of phagophores with vacuoles [
15]. Most likely, the other components of the Ypt7 module would also positively regulate this fusion process.
Negative regulators during fusion of stuffs to vacuoles were seldom reported, but such regulators do exist. The VTC complex proteins negatively regulate the entry of misfolded GPI-anchored proteins in
pep4∆ cells into vacuoles during microautophagy [
24]. The VTC complex proteins also negatively regulate the entry of phagophores into vacuoles in
vps21∆ cells as the further depletion of either four out of five proteins in the VTC complex promoted the entry of phagophores into vacuoles and increased phagophore degradation [
15]. If the depletion of the VTC complex protein also promoted the phagophores in ESCRT mutant
snf7∆ cells to enter vacuoles under nitrogen starvation, we expected that there would be more phagophores inside vacuoles in
snf7∆
vtc4∆ cells than in
snf7∆ cells. We compared phagophore localization and degradation in
snf7∆ and
snf7∆
vtc4∆ cells in this study. The
vps21∆ and
vps21∆
vtc4∆ cells were included as control cells. Our results showed that the percentage of cells with accumulated phagophores on vacuolar membranes in
vps21∆ and
snf7∆ cells significantly decreased when Vtc4 was depleted, no matter whether GFP-Atg8 or mCherry-Atg8 were used to label the phagophores (
Figure 1 and
Figure 3). The mean percentage of GFP degraded from GFP-Atg8 in
vps21∆
vtc4∆ and
snf7∆
vtc4∆ cells was always higher than that in
vps21∆ and
snf7∆ cells although without significance (
Figure 2), which may be due to the relatively low GFP-Atg8 degradation in mutants due to a possible decrease in hydrolysis ability. Alternatively, the decreased degradation of VTC complex proteins through vacuolar proteases in ESCRT mutant cells could not be excluded, as some Vtc5 is rerouted in the vacuolar lumen by ESCRT and immediately undergoes protease degradation [
34]. If the degradation of VTC complex proteins in
snf7∆ cells was decreased, there might be more VTC complex proteins to inhibit the entry of phagophores into vacuoles and be degraded. In addition, the degradation of GFP-Atg8 is highly dependent on the cell density, availability of vacuolar hydrolases and the nitrogen starvation time in
vps21∆ and ESCRT mutant cells (Figures 1C–F, S1C and S6B,D in [
15]). We do not know how these mutations affect the dynamic ability of hydrolases or the degradation of VTC complex proteins through vacuolar proteases. We always control the experiment conditions to be the same, with the strain as the only variation in the same experiment. Under this situation, we can strictly compare the difference in GFP-Atg8 degradation between
vps21∆ and
vps21∆
vtc4∆ cells or
snf7∆ and
snf7∆
vtc4∆ cells at the same nitrogen starvation time, as shown in
Figure 2. However, when the changes in GFP-Atg8 degradation are minor and the sensitivity of ImageJ measurement always has limitations, the difference in the calculated percentage of GFP may be not significant between
vps21∆ and
vps21∆
vtc4∆ cells or
snf7∆ and
snf7∆
vtc4∆ cells. All these data are not related to florescence degradation or excessive exposure of the film. Finally, the molecular mechanism of Vtc4 depletion in promoting phagophore entry into vacuoles is still unclear, which needs further investigation.
ESCRT complex is a multiple subunit complex. ESCRT-III subunit Snf7 and AAA-ATPase Vps4 are the most frequent and well-studied subunits for their roles in vesicle trafficking and autophagy [
35]. In our previous study in [
12], we already showed that most ESCRT mutant cells, especially
snf7∆ and
vps4∆ cells, had similar and the strongest phenotypes for phagophore accumulation. We confirmed that the phagophore accumulation phenotypes in
snf7∆ and
vps4∆ cells are quite similar either under short or prolonged periods of nitrogen starvation (
Figures S1 and S2 and [
12,
15,
36]). Therefore, the study contents for
snf7∆ cells in this study are most likely representative for ESCRT mutant cells. Besides Vtc4, depletion of Vtc1, 2 or 5 also promoted the entry of phagophores into vacuoles in
vps21∆ cells at similar levels [
15]. The examinations of Vtc4 depletion in
snf7∆ cells most likely will be representative for the examinations of the depletion of any VTC complex proteins (Vtc1, 2, 4 and 5) in ESCRT mutant cells. We did not examine the effects of depletion of other VTC complex proteins in
snf7∆ cells, and we also did not examine the depletion of any VTC complex proteins (Vtc1, 2, 4 and 5) in
vps4∆ cells. If these experiments were performed, it is expected that they will generate similar results and conclusions about Vtc4 depletion in
snf7∆ cells, although this needs future experiments to support it. Mammalian ESCRT complex subunits are also required for phagophore closure, as phagophores accumulate in cytosol when the key ESCRT complex subunits are absent [
36]. The size of the mammalian phagophore or autophagosome is relatively bigger than the size of the lysosome. There is no chance for mammalian phagophores or autophagosomes to enter lysosomes. In contrast, multiple mammalian lysosomes fuse with a phagophore or autophagosome to provide hydrolases to degrade them for recycling [
37]. The similar positive fusion regulators as in yeast cells for phagophore or autophagosome–vacuole fusion are required for autophagosome–lysosome fusion in mammalian cells [
38]. However, it is unclear whether there are negative regulators like VTC complex proteins in mammalian cells to promote the phagophore–lysosome fusion.
Our previous study showed that the entry of phagophores in
vps21∆ cells was promoted by the depletion of VTC complex proteins [
15]. This study further confirmed and broadened the promotion effect of depletion of the VTC complex protein on phagophore entry into vacuoles in yeast by using ESCRT mutant. These findings not only expand the functions of VTC complex but also provide information for the regulation of phagophores into vacuoles. Vtc4 depletion promotes the entry of phagophores into vacuoles in
vps21∆ or
snf7∆ cells; however, it is unclear whether Vps21 or Snf7 and Vtc4 act in a dependent way. Most likely, the accumulation of phagophores outside vacuoles by Vps21 or Snf7 depletion under nitrogen starvation provides a convenient way to study the effect of Vtc4 depletion to promote the phagophore entry. Vtc4 depletion did not have obvious promotion effect if the core four strains in
Figure 1B were not starved to induce autophagy to form phagophores. We also noticed that the amount of GFP-Atg8 degradation in
vtc4∆ cells was also more than that in wild-type cells in the mean value in
Figure 2. We do not think that Vps21 and Vtc4 or Snf7 and Vtc4 act in a dependent way. We proposed that the VTC complex may have more roles to negatively regulate other cargoes outside or on vacuolar membranes to enter vacuoles in yeast. However, it is unclear whether such a mechanism also exists in mammalian cells.



{kind=link}
{kind=link}
{kind=link}