
Towards a High-Affinity Peptidomimetic Targeting Proliferating Cell Nuclear Antigen from Aspergillus fumigatus

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Peptides
2.1.1. Peptide Synthesis by Fmoc SPPS
2.2. Expression of Recombinant afumPCNA
2.3. Purification of Recombinant afumPCNA
2.4. Surface Plasmon Resonance Protocol
2.5. Protein-Peptide Co-Crystallization Experiments
2.6. Computational Modelling
3. Results
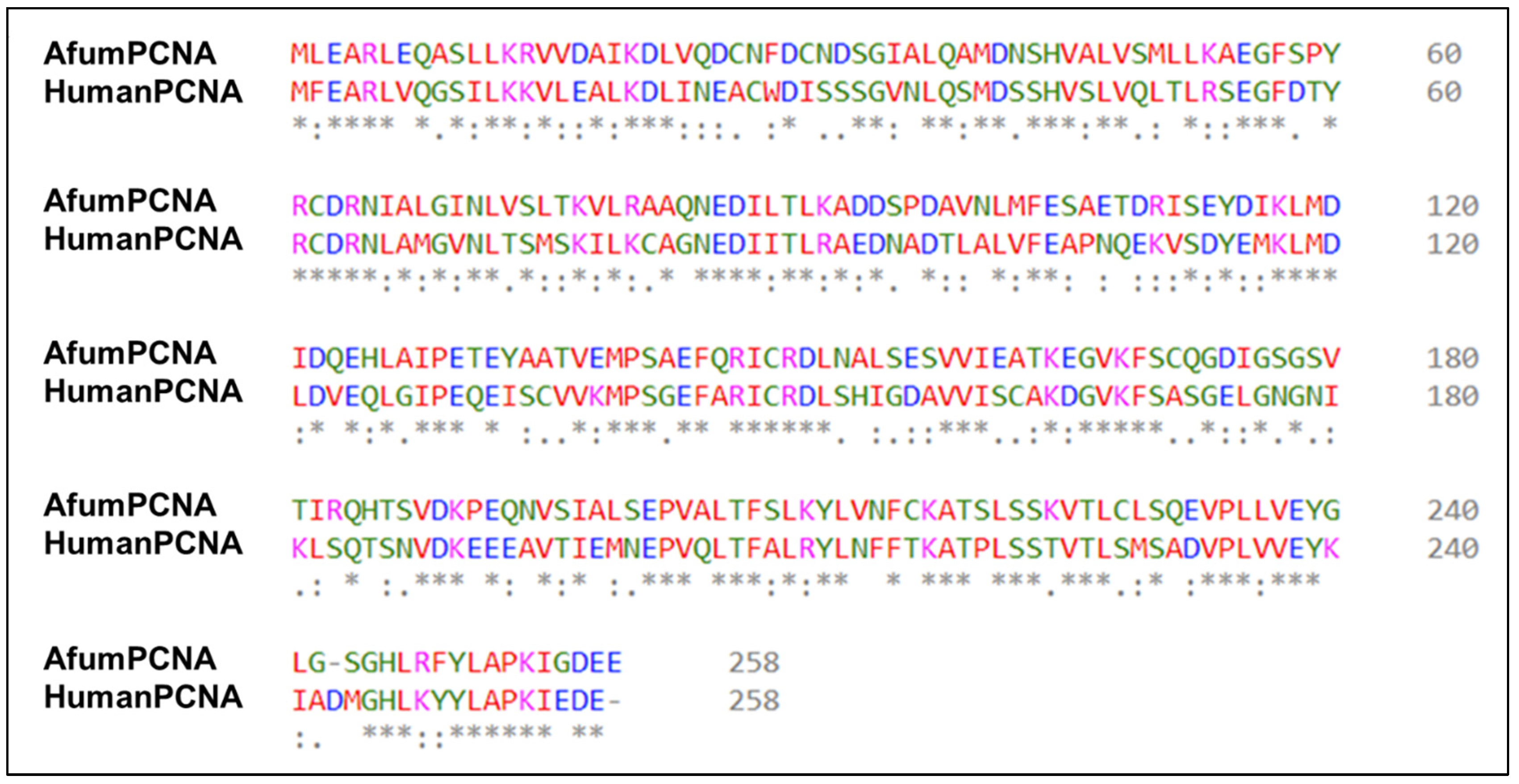
3.1. A p21 Peptide Library Interacts with afumPCNA in a Similar Trend Compared to hPCNA
3.2. Fungal Protein PIP-Box Candidate Discovery Displays a Surprising afumPCNA Interaction
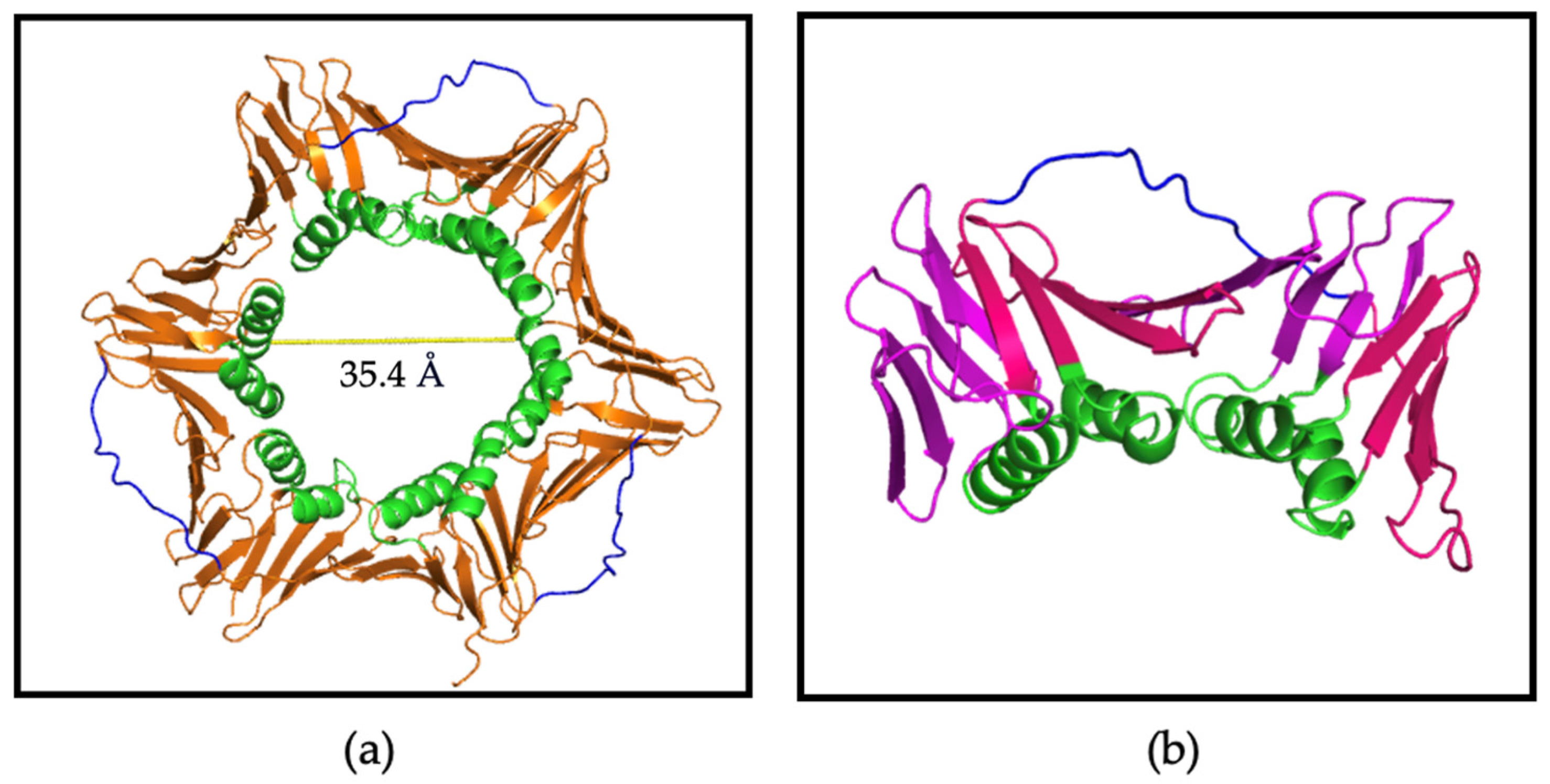
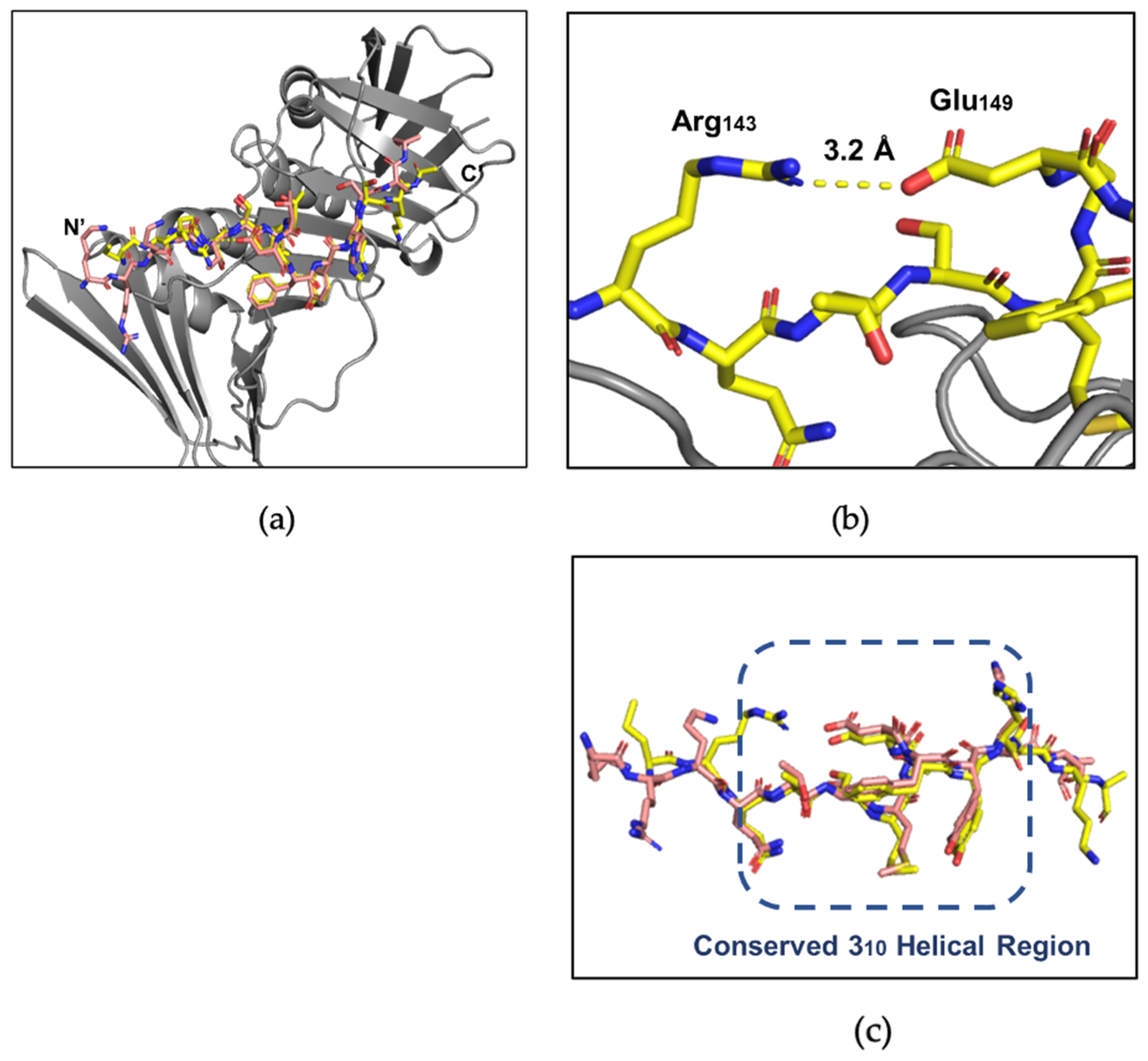
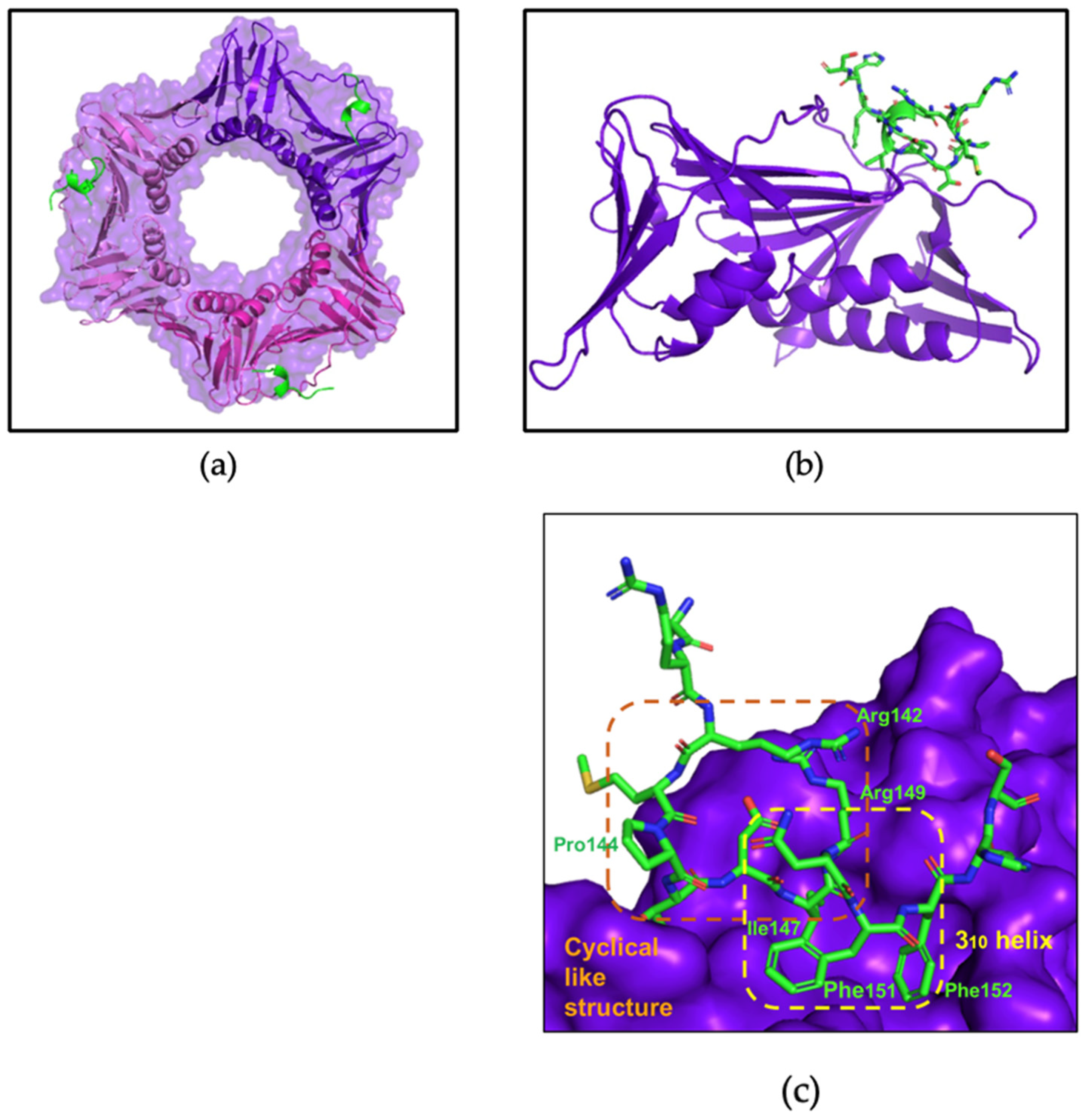
3.3. X-ray Crystallography Study of the p21µ Peptide Bound to afumPCNA
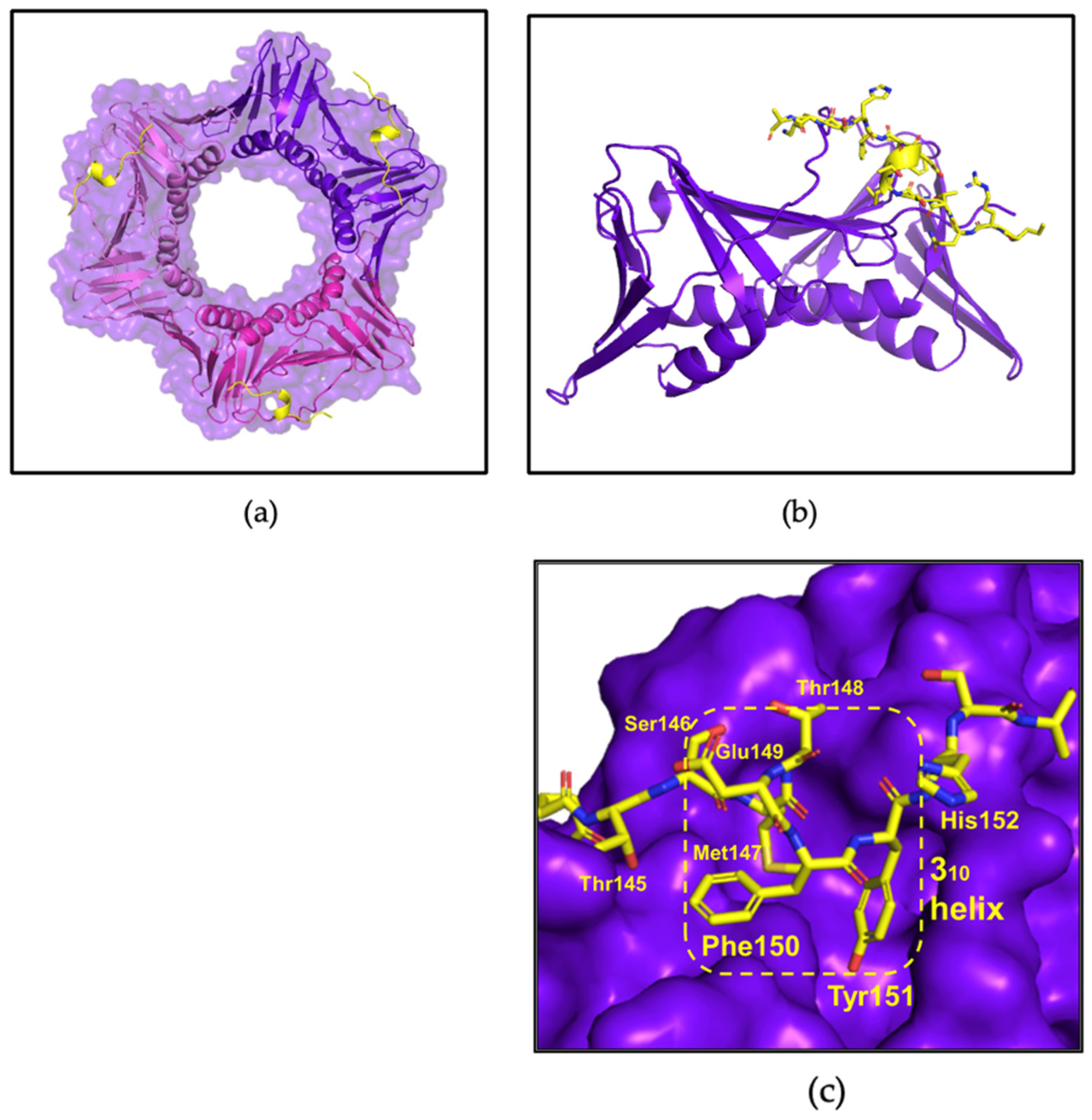
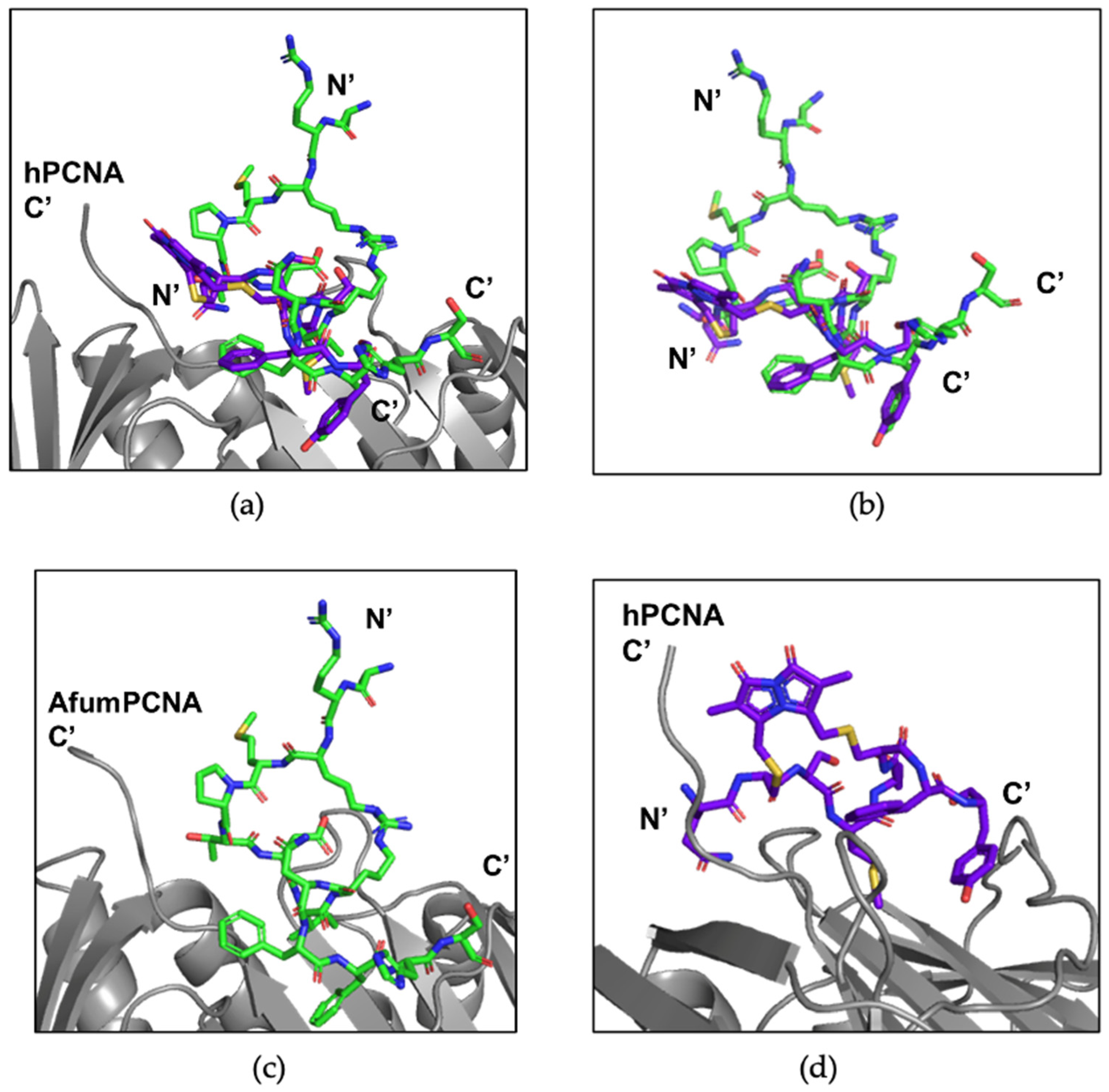
3.4. X-ray Crystallography Study of afumPCNA and p21µ-afumRFC Reveals a Ring-like Structure
4. Discussion
4.1. The p21 Peptide Library Interacts with afumPCNA with Similar Affinity and Structural Trends as hPCNA
4.2. Fungal Protein Replication Factor C PIP-Box Candidate Pepide Has a High Affinity for afumPCNA
4.3. The p21μafumRFC Peptide Has a Unique Structure That Could Be Exploited for an Antifungal Treatment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bitar, D.; Lortholary, O.; Le Strat, Y.; Nicolau, J.; Coignard, B.; Tattevin, P.; Che, D.; Dromer, F. Population-based analysis of invasive fungal infections, France, 2001–2010. Emerg. Infect. Dis. 2014, 20, 1149–1155. [Google Scholar] [CrossRef]
- McNeil, M.; Nash, S.L.; Hajjeh, R.A.; Phelan, M.A.; Conn, L.A.; Plikaytis, B.D.; Warnock, D.W. Trends in mortality due to invasive mycotic diseases in the United States, 1980–1997. Clin. Infect. Dis. 2001, 33, 641–647. [Google Scholar] [CrossRef]
- Latgé, J.-P.; Chamilos, G. Aspergillus fumigatus and Aspergillosis in 2019. Clin. Microbiol. Rev. 2019, 33, 00140-18. [Google Scholar] [CrossRef]
- Rhodes, J.; Abdolrasouli, A.; Dunne, K.; Sewell, T.R.; Zhang, Y.; Ballard, E.; Brackin, A.P.; van Rhijn, N.; Chown, H.; Tsitsopoulou, A.; et al. Population genomics confirms acquisition of drug-resistant Aspergillus fumigatus infection by humans from the environment. Nat. Microbiol. 2022, 7, 663–674. [Google Scholar] [CrossRef]
- Yoon, H.; Choi, H.Y.; Kim, Y.K.; Song, Y.J.; Ki, M. Prevalence of fungal infections using National Health Insurance data from 2009–2013, South Korea. Epidemiol. Health 2014, 36, e2014017. [Google Scholar] [CrossRef]
- Van de Veerdonk, F.L.; Gresnigt, M.S.; Romani, L.; Netea, M.G.; Latgé, J.-P. Aspergillus fumigatus morphology and dynamic host interactions. Nat. Rev. Microbiol. 2017, 15, 661–674. [Google Scholar] [CrossRef]
- Gallien, S.; Fournier, S.; Porcher, R.; Bottero, J.; Ribaud, P.; Sulahian, A.; Molina, J.M. Therapeutic outcome and prognostic factors of invasive aspergillosis in an infectious disease department: A review of 34 cases. Infection 2008, 36, 533–538. [Google Scholar] [CrossRef]
- Sugui, J.; Kwon-Chung, K.J.; Juvvadi, P.R.; Latge, J.P.; Steinbach, W.J. Aspergillus fumigatus and related species. Cold Spring Harb Perspect. Med. 2015, 5, a019786. [Google Scholar] [CrossRef]
- Steinbach, W. Are we there yet? Recent progress in the molecular diagnosis and novel antifungal targeting of Aspergillus fumigatus and invasive aspergillosis. PLoS Pathog. 2013, 9, e1003642. [Google Scholar] [CrossRef]
- Arastehfar, A.; Carvalho, A.; Houbraken, J.; Lombardi, L.; Garcia-Rubio, R.; Jenks, J.D.; Rivero-Menendez, O.; Aljohani, R.; Jacobsen, I.D.; Berman, J.; et al. Aspergillus fumigatus and aspergillosis: From basics to clinics. Stud. Mycol. 2021, 100, 100115. [Google Scholar] [CrossRef]
- Ben-Ami, R.; Lewis, R.E.; Kontoyiannis, D.P. Enemy of the (immunosuppressed) state: An update on the pathogenesis of Aspergillus fumigatus infection. Br. J. Haematol. 2010, 150, 406–417. [Google Scholar] [CrossRef]
- Resendiz Sharpe, A.; Lagrou, K.; Meis, J.F.; Chowdhary, A.; Lockhart, S.R.; Verweij, P.E.; ISHAM/ECMM Aspergillus Resistance Surveillance Working Group. Triazole resistance surveillance in Aspergillus fumigatus. Med. Mycol. 2018, 56, S83–S92. [Google Scholar] [CrossRef] [PubMed]
- Edouarzin, E.; Horn, C.; Paudyal, A.; Zhang, C.; Lu, J.; Tong, Z.; Giaever, G.; Nislow, C.; Veerapandian, R.; Hua, D.H.; et al. Broad-spectrum antifungal activities and mechanism of drimane sesquiterpenoids. Microb. Cell 2020, 7, 146–159. [Google Scholar] [CrossRef]
- Marshall, A.C.; Kroker, A.J.; Murray, L.A.; Gronthos, K.; Rajapaksha, H.; Wegener, K.L.; Bruning, J.B. Structure of the sliding clamp from the fungal pathogen Aspergillus fumigatus (AfumPCNA) and interactions with human p21. FEBS J. 2017, 284, 985–1002. [Google Scholar] [CrossRef] [PubMed]
- Maga, G.; Hubscher, U. Proliferating cell nuclear antigen (PCNA): A dancer with many partners. J. Cell Sci. 2003, 116, 3051–3060. [Google Scholar] [CrossRef]
- Sakakura, C.; Hagiwara, A.; Tsujimoto, H.; Ozaki, K.; Sakakibara, T.; Oyama, T.; Takahashi, T. The anti-proliferative effect of proliferating cell nuclear antigen-specific antisense oligonucleotides on human gastric cancer cell lines. Surg. Today 1995, 25, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.; Lee, H. Characterization of proliferating cell nuclear antigen (PCNA) isoforms in normal and cancer cells: There is no cancer-associated form of PCNA. FEBS Lett. 2007, 528, 4917–4920. [Google Scholar] [CrossRef] [PubMed]
- Horsfall, A.J.; Vandborg, B.A.; Kowalczyk, W.; Chav, T.; Scanlon, D.B.; Abell, A.D.; Bruning, J.B. Unlocking the PIP-box: A peptide library reveals interactions that drive high-affinity binding to human PCNA. J. Biol. Chem. 2021, 296, 100773. [Google Scholar] [CrossRef]
- Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.2r3pre; Schrodinger LLC: New York, NY, USA, 2015.
- Warbrick, E. PCNA binding through a conserved motif. Bioessays 1998, 20, 195–199. [Google Scholar] [CrossRef]
- Gulbis, J.M.; Kelman, Z.; Hurwitz, J.; O’Donnell, M.; Kuriyan, J. Structure of the C terminal region of p21(WAF1/CIP1) com- plexed with human PCNA. Cell 1996, 87, 297–306. [Google Scholar] [CrossRef]
- Prestel, A.; Wichmann, N.; Martins, J.M.; Marabini, R.; Kassem, N.; Broendum, S.S.; Otterlei, M.; Nielsen, O.; Willemoes, M.; Ploug, M.; et al. The PCNA interaction motifs revisited: Thinking outside the PIP-box. Cell. Mol. Life Sci. 2019, 76, 4923–4943. [Google Scholar] [CrossRef] [PubMed]
- Vandborg, B.; Holroyd, D.L.; Pukala, T.; Bruning, J.B. Production of recombinant human proliferating cellular nuclear antigen (PCNA) for structural and biophysical characterization. Protein Expr. Purif. 2023, 212, 106353. [Google Scholar] [CrossRef] [PubMed]
- Anthis, N.J.; Clore, G.M. Sequence-specific determination of protein and peptide concentrations by absorbance at 205 nm. Protein Sci. 2013, 22, 851–858. [Google Scholar] [CrossRef]
- Kabsch, W. XDS (X-ray detector software). Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Potterton, E.; Briggs, P.J.; Turkenburg, M.G.W.; Dodson, E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 1131–1137. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storonia, L.C.; Reada, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.Refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Phenix: Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in phenix. Acta Crystallogr. D Biol. Crystallogr. 2019, 75, 861–877. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Horsfall, A.; Abell, A.; Bruning, J. Targeting PCNA with peptide mimetics for therapeutic purposes. ChemBioChem 2019, 21, 442–450. [Google Scholar] [CrossRef]
- Boehm, E.; Washington, M.T. R.I.P. to the PIP: PCNA-binding motif no longer considered specific. BioEssays 2016, 38, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, P.G.; Ashweta, S.; Dehua, P. Understanding Cell Penetration of Cyclic Peptides. Chem. Rev. 2019, 119, 10241–10287. [Google Scholar] [CrossRef] [PubMed]
- Wegener, K.L.; McGrath Amy, E.; Dixon Nicholas, E.; Oakley Aaron, J.; Scanlon Denis, B.; Abell Andrew, D.; Bruning John, B. Rational Design of a 310 -Helical PIP-Box Mimetic Targeting hPCNA, the Human Sliding Clamp. Eur. J. Chem. 2018, 24, 11325–11331. [Google Scholar] [CrossRef]
- Horsfall, A.J.; Vandborg, B.A.; Kikhtyak, Z.; Scanlon, D.B.; Tilley, W.D.; Hickey, T.E.; Bruning, J.B.; Abell, A.D. A cell permeable bimane-constrained PCNA-interacting peptide. RSC Chem. Biol. 2021, 2, 1499–1508. [Google Scholar] [CrossRef]
- Gong, Z.; Karlsson, A.J. Translocation of cell-penetrating peptides into Candida fungal pathogens. Protein Sci. 2017, 26, 1714–1725. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | afumPCNA Binding Affinity KD (nM) ± SD (nM) | Human PCNA Binding Affinity KD (nM) ± SD (nM) [18] |
|---|---|---|---|
| p21(139–160) | 139GRKRR QTSMTDFY HSKRRLIFS160 | 69.7 ± 20.2 | 4.3 ± 1.3 |
| p21μ | 141KRR QTSMTDFY HSKR155 | 265.1 ± 5.9 | 12.3 ± 0.5 |
| p21μ-RD2 | 141KRR QTRITEYF HSKR155 | 20.3 ± 6.8 | 1.1 ± 0.3 |
| p21μ-Q144M | 141KRR MTSMTDFY HSKR155 | 41,400 ± 0.8 | 1544 ± 159 |
| p21μ-T145K | 141KRR QKSMTDFY HSKR155 | 10,000 ± 0.7 | 98 ± 10.8 |
| p21μ-T145D | 141KRR QDSMTDFY HSKR155 | 4100 ± 0.31 | 714 ± 30.4 |
| p21μ-S146R | 141KRR QTRMTDFY HSKR155 | 64.4 ± 18.4 | 4.3 ± 1.3 |
| p21μ-M147L | 141KRR QTSLTDFY HSKR155 | 382 ± 51.0 | 20.5 ± 1.7 |
| p21μ-M147I | 141KRR QTSITDFY HSKR155 | 37.1 ± 7.8 | 11.1 ± 0.3 |
| p21μ-D149E | 141KRR QTSMTEFY HSKR155 | 400.7 ± 45.6 | 12.7 ± 1.4 |
| p21μ-F150Y | 141KRR QTSMTDYY HSKR155 | 75.2 ± 18.9 | 20.2 ± 0.4 |
| p21μ-Y151F | 141KRR QTSMTDFF HSKR155 | 167.2 ± 19.1 | 10.6 ± 1.5 |
| p21μ-FY150151YF | 141KRR QTSMTDYF HSKR155 | 96.4 ± 19.6 | 2.2 ± 0.5 |
| Protein Name | Homo Sapiens PIP-Box Sequence | Aspergillus fumigatus PIP-Box Sequence Candidates |
|---|---|---|
| FEN1 | QGRLDDFF | QSRLEGFF |
| RFC1 | MDIRKFF | MPTDIRNFF |
| DNA Polymerase POLD3 | QVSITGFF | QKELSRFD |
| DNA Ligase | QRSIMSFF | QRVRSIASFF |
| Name | Sequence | afumPCNA Binding Affinity KD (nM) ± KD SD (nM) | Human PCNA Binding Affinity KD (nM) ± KD SD (nM) |
|---|---|---|---|
| p21μafumDNALIG | 141KRRQRVRSIASFFHSKR157 | 458 ± 117.77 | - |
| p21μafumDNAPOL | 141KRRQKELSRFDFHSKR156 | 659.3 ± 105.8 | - |
| p21μafumFEN1 | 141KRRQSRLEGFFHSKR155 | 713 ± 56.9 | - |
| p21μafumRFC | 141KRRMPTDIRNFFHSKR156 | 94.84 ± 8.76 | 295 ± 6.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vandborg, B.C.; Horsfall, A.J.; Pederick, J.L.; Abell, A.D.; Bruning, J.B. Towards a High-Affinity Peptidomimetic Targeting Proliferating Cell Nuclear Antigen from Aspergillus fumigatus. J. Fungi 2023, 9, 1098. https://doi.org/10.3390/jof9111098
Vandborg BC, Horsfall AJ, Pederick JL, Abell AD, Bruning JB. Towards a High-Affinity Peptidomimetic Targeting Proliferating Cell Nuclear Antigen from Aspergillus fumigatus. Journal of Fungi. 2023; 9(11):1098. https://doi.org/10.3390/jof9111098
Chicago/Turabian StyleVandborg, Bethiney C., Aimee J. Horsfall, Jordan L. Pederick, Andrew D. Abell, and John B. Bruning. 2023. "Towards a High-Affinity Peptidomimetic Targeting Proliferating Cell Nuclear Antigen from Aspergillus fumigatus" Journal of Fungi 9, no. 11: 1098. https://doi.org/10.3390/jof9111098
APA StyleVandborg, B. C., Horsfall, A. J., Pederick, J. L., Abell, A. D., & Bruning, J. B. (2023). Towards a High-Affinity Peptidomimetic Targeting Proliferating Cell Nuclear Antigen from Aspergillus fumigatus. Journal of Fungi, 9(11), 1098. https://doi.org/10.3390/jof9111098