
A Landscape of the Genomic Structure of Cryptococcus neoformans in Colombian Isolates
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Whole Genome Sequencing
2.3. Phylogenomic Inferences
2.4. Gene Copy Number Variations
2.5. Variant Prediction Calling, SNP Filtering, and Genetic Diversity Analysis
3. Results
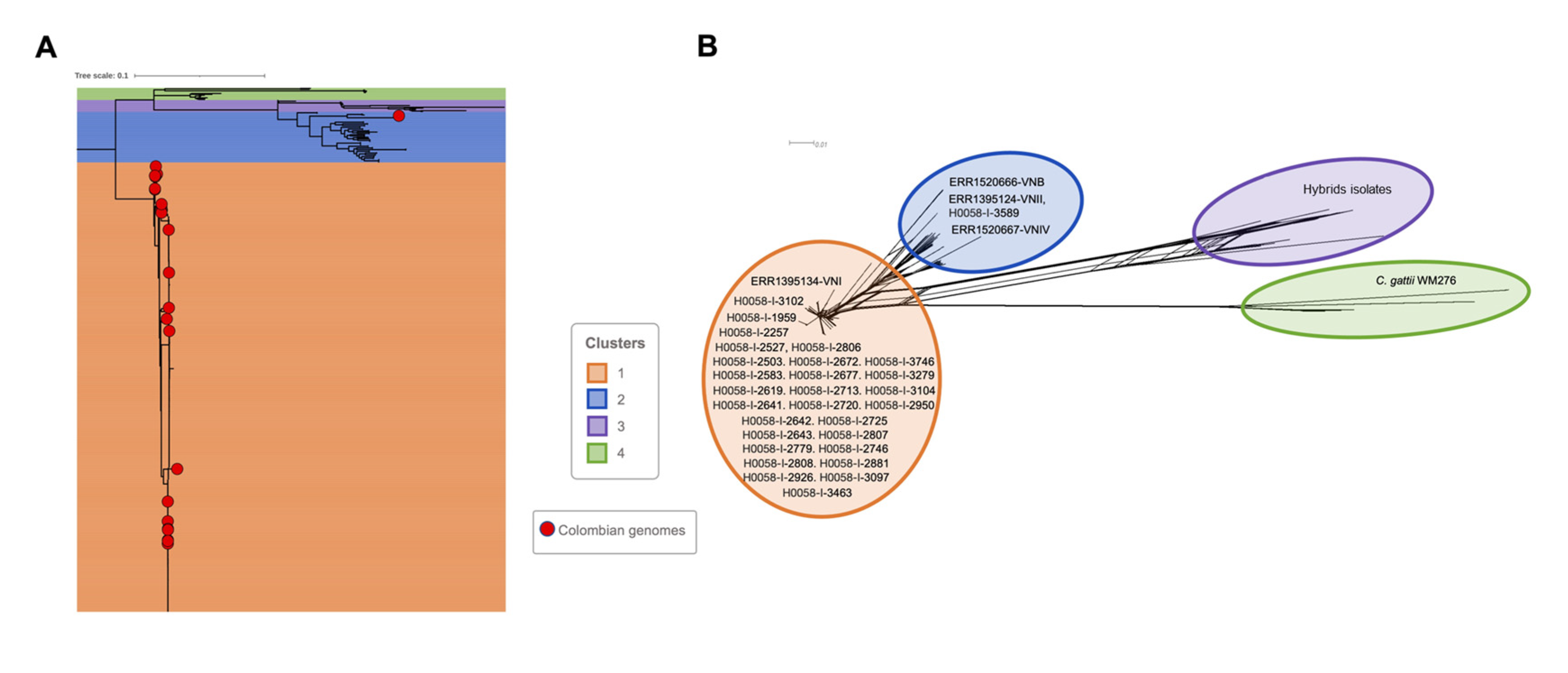
3.1. Phylogenomic Analysis
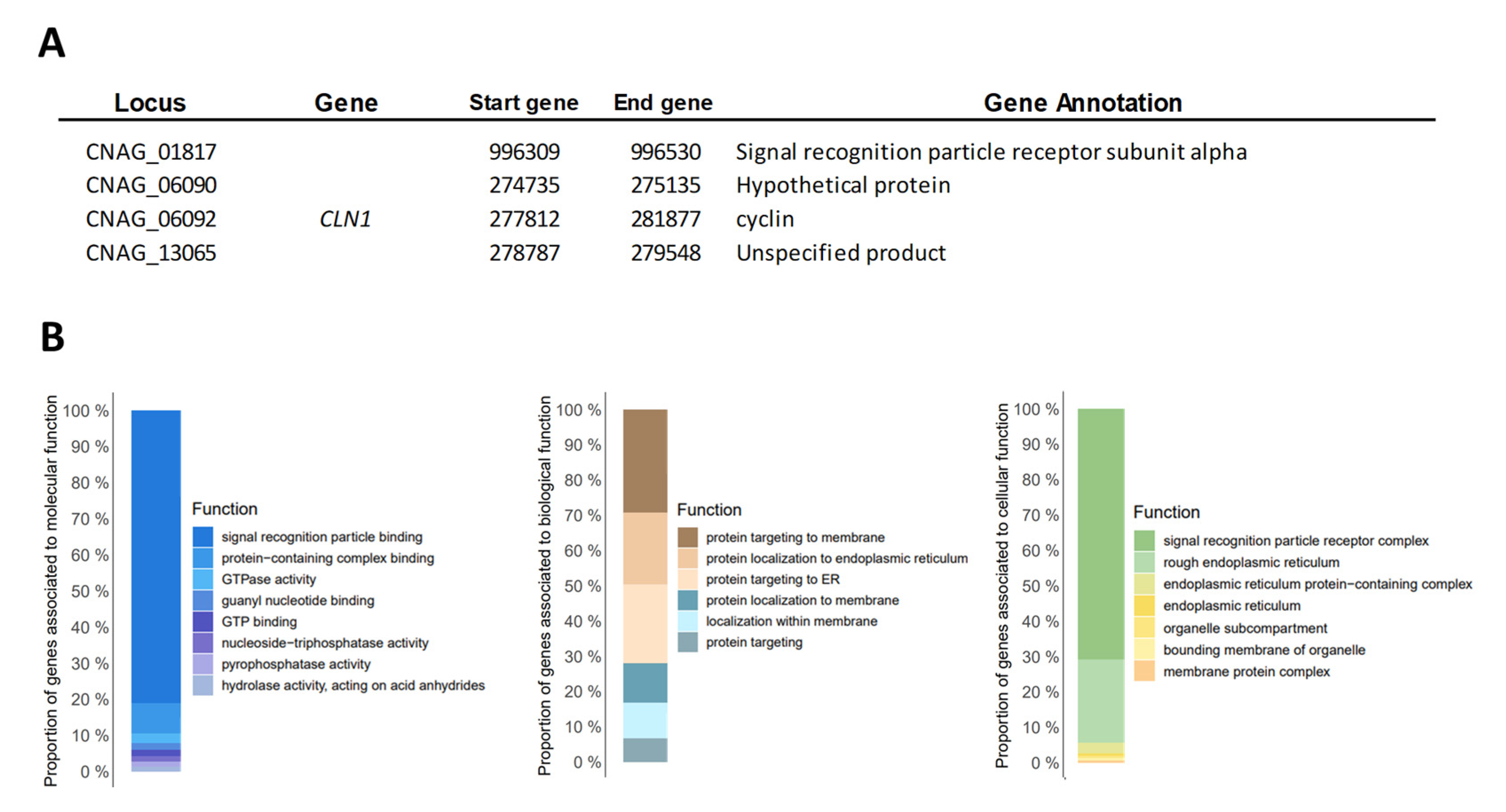
3.2. Gene Copy Number Variation Analysis (CNV)
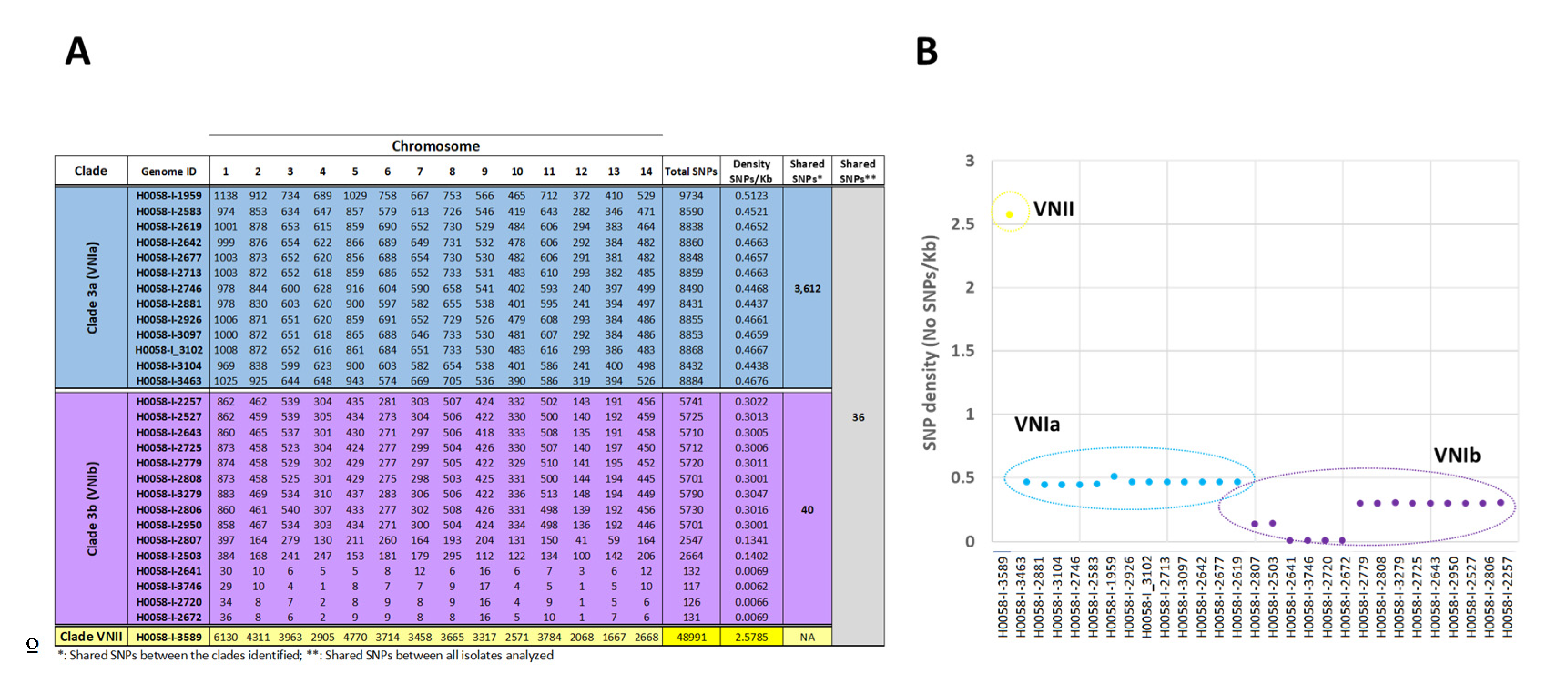
3.3. Single Nucleotide Polymorphisms Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wiederhold, N.P. Emerging Fungal Infections: New Species, New Names, and Antifungal Resistance. Clin. Chem. 2021, 68, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Rajasingham, R.; Smith, R.M.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.M.; Denning, D.W.; Loyse, A.; Boulware, D.R. Global burden of disease of HIV-associated cryptococcal meningitis: An updated analysis. Lancet Infect. Dis. 2017, 17, 873–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firacative, C.; Meyer, W.; Castaneda, E. Cryptococcus neoformans and Cryptococcus gattii Species Complexes in Latin America: A Map of Molecular Types, Genotypic Diversity, and Antifungal Susceptibility as Reported by the Latin American Cryptococcal Study Group. J. Fungi 2021, 7, 282. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, M. Global Molecular Epidemiology of Cryptococcus neoformans and Cryptococcus gattii: An Atlas of the Molecular Types. Scientifica 2013, 2013, 675213. [Google Scholar] [CrossRef] [Green Version]
- Escandon, P.; Lizarazo, J.; Agudelo, C.I.; Castaneda, E. Cryptococcosis in Colombia: Compilation and Analysis of Data from Laboratory-Based Surveillance. J. Fungi 2018, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- Beardsley, J.; Dao, A.; Keighley, C.; Garnham, K.; Halliday, C.; Chen, S.; Sorrell, T. What’s New in Cryptococcus gattii: From Bench to Bedside and Beyond. J. Fungi 2023, 9, 41. [Google Scholar] [CrossRef]
- Hagen, F.; Khayhan, K.; Theelen, B.; Kolecka, A.; Polacheck, I.; Sionov, E.; Falk, R.; Parnmen, S.; Lumbsch, H.T.; Boekhout, T. Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet. Biol. 2015, 78, 16–48. [Google Scholar] [CrossRef] [Green Version]
- Edwards, H.M.; Cogliati, M.; Kwenda, G.; Fisher, M.C. The need for environmental surveillance to understand the ecology, epidemiology and impact of Cryptococcus infection in Africa. FEMS Microbiol. Ecol. 2021, 97, fiab093. [Google Scholar] [CrossRef]
- Springer, D.J.; Billmyre, R.B.; Filler, E.E.; Voelz, K.; Pursall, R.; Mieczkowski, P.A.; Larsen, R.A.; Dietrich, F.S.; May, R.C.; Filler, S.G.; et al. Cryptococcus gattii VGIII isolates causing infections in HIV/AIDS patients in Southern California: Identification of the local environmental source as arboreal. PLoS Pathog. 2014, 10, e1004285. [Google Scholar] [CrossRef] [Green Version]
- Farrer, R.A.; Chang, M.; Davis, M.J.; van Dorp, L.; Yang, D.H.; Shea, T.; Sewell, T.R.; Meyer, W.; Balloux, F.; Edwards, H.M.; et al. A New Lineage of Cryptococcus gattii (VGV) Discovered in the Central Zambezian Miombo Woodlands. mBio 2019, 10, e02306-19. [Google Scholar] [CrossRef]
- Idnurm, A.; Bahn, Y.S.; Nielsen, K.; Lin, X.; Fraser, J.A.; Heitman, J. Deciphering the model pathogenic fungus Cryptococcus neoformans. Nat. Rev. Microbiol. 2005, 3, 753–764. [Google Scholar] [CrossRef]
- Viviani, M.A.; Cogliati, M.; Esposto, M.C.; Lemmer, K.; Tintelnot, K.; Colom Valiente, M.F.; Swinne, D.; Velegraki, A.; Velho, R. European Confederation of Medical Mycology Cryptococcosis Working G. Molecular analysis of 311 Cryptococcus neoformans isolates from a 30-month ECMM survey of Cryptococcosis in Europe. FEMS Yeast Res. 2006, 6, 614–619. [Google Scholar] [CrossRef] [Green Version]
- Kwon-Chung, K.J.; Fraser, J.A.; Doering, T.L.; Wang, Z.; Janbon, G.; Idnurm, A.; Bahn, Y.S. Cryptococcus neoformans and Cryptococcus gattii, the etiologic agents of Cryptococcosis. Cold Spring Harb. Perspect. Med. 2014, 4, a019760. [Google Scholar] [CrossRef] [Green Version]
- Serna-Espinosa, B.N.; Guzman-Sanabria, D.; Forero-Castro, M.; Escandon, P.; Sanchez-Quitian, Z.A. Environmental Status of Cryptococcus neoformans and Cryptococcus gattii in Colombia. J. Fungi 2021, 7, 410. [Google Scholar] [CrossRef]
- Anacona, C.; Gonzalez, C.F.; Vasquez, A.L.; Escandon, P. First isolation and molecular characterization of Cryptococcus neoformans var. grubii in excreta of birds in the urban perimeter of the Municipality of Popayan, Colombia. Rev. Iberoam. Micol. 2018, 35, 123–129. [Google Scholar] [CrossRef]
- van Rhijn, N.; Bromley, M. The Consequences of Our Changing Environment on Life Threatening and Debilitating Fungal Diseases in Humans. J. Fungi 2021, 7, 367. [Google Scholar] [CrossRef]
- Nnadi, N.E.; Carter, D.A. Climate change and the emergence of fungal pathogens. PLoS Pathog. 2021, 17, e1009503. [Google Scholar] [CrossRef]
- Saracli, M.A.; Yildiran, S.T.; Sener, K.; Gonlum, A.; Doganci, L.; Keller, S.M.; Wickes, B.L. Genotyping of Turkish environmental Cryptococcus neoformans var. neoformans isolates by pulsed field gel electrophoresis and mating type. Mycoses 2006, 49, 124–129. [Google Scholar] [CrossRef]
- Ngamwongsatit, P.; Sukroongreung, S.; Nilakul, C.; Prachayasittikul, V.; Tantimavanich, S. Electrophoretic karyotypes of C. neoformans serotype A recovered from Thai patients with AIDS. Mycopathologia 2005, 159, 189–197. [Google Scholar] [CrossRef]
- Munoz, M.; Camargo, M.; Ramirez, J.D. Estimating the Intra-taxa Diversity, Population Genetic Structure, and Evolutionary Pathways of Cryptococcus neoformans and Cryptococcus gattii. Front. Genet. 2018, 9, 148. [Google Scholar] [CrossRef]
- Velez, N.; Vega-Vela, N.; Munoz, M.; Gomez, P.; Escandon, P.; Ramirez, J.D.; Zaragoza, O.; Monteoliva Diaz, L.; Parra-Giraldo, C.M. Deciphering the Association among Pathogenicity, Production and Polymorphisms of Capsule/Melanin in Clinical Isolates of Cryptococcus neoformans var. grubii VNI. J. Fungi 2022, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, C.A.; Rhodes, J.; Desjardins, C.A. Advances in Cryptococcus genomics: Insights into the evolution of pathogenesis. Mem Inst. Oswaldo Cruz 2018, 113, e170473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, J.; Beale, M.A.; Vanhove, M.; Jarvis, J.N.; Kannambath, S.; Simpson, J.A.; Ryan, A.; Meintjes, G.; Harrison, T.S.; Fisher, M.C.; et al. A Population Genomics Approach to Assessing the Genetic Basis of Within-Host Microevolution Underlying Recurrent Cryptococcal Meningitis Infection. G3 Genes Genomes Genet. 2017, 7, 1165–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billmyre, R.B.; Clancey, S.A.; Heitman, J. Natural mismatch repair mutations mediate phenotypic diversity and drug resistance in Cryptococcus deuterogattii. Elife 2017, 6, e28802. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, C.A.; Giamberardino, C.; Sykes, S.M.; Yu, C.H.; Tenor, J.L.; Chen, Y.; Yang, T.; Jones, A.M.; Sun, S.; Haverkamp, M.R.; et al. Population genomics and the evolution of virulence in the fungal pathogen Cryptococcus neoformans. Genome Res. 2017, 27, 1207–1219. [Google Scholar] [CrossRef] [Green Version]
- Hong, N.; Chen, M.; Xu, J. Molecular Markers Reveal Epidemiological Patterns and Evolutionary Histories of the Human Pathogenic Cryptococcus. Front. Cell. Infect. Microbiol. 2021, 11, 683670. [Google Scholar] [CrossRef]
- Castanheira, M.; Deshpande, L.M.; Davis, A.P.; Rhomberg, P.R.; Pfaller, M.A. Monitoring Antifungal Resistance in a Global Collection of Invasive Yeasts and Molds: Application of CLSI Epidemiological Cutoff Values and Whole-Genome Sequencing Analysis for Detection of Azole Resistance in Candida albicans. Antimicrob. Agents Chemother. 2017, 61, e00906-17. [Google Scholar] [CrossRef] [Green Version]
- Billmyre, R.B.; Croll, D.; Li, W.; Mieczkowski, P.; Carter, D.A.; Cuomo, C.A.; Kronstad, J.W.; Heitman, J. Highly recombinant VGII Cryptococcus gattii population develops clonal outbreak clusters through both sexual macroevolution and asexual microevolution. mBio 2014, 5, e01494-14. [Google Scholar] [CrossRef] [Green Version]
- Samarasinghe, H.; Xu, J. Hybrids and hybridization in the Cryptococcus neoformans and Cryptococcus gattii species complexes. Infect. Genet Evol. 2018, 66, 245–255. [Google Scholar] [CrossRef]
- Ashton, P.M.; Thanh, L.T.; Trieu, P.H.; Van Anh, D.; Trinh, N.M.; Beardsley, J.; Kibengo, F.; Chierakul, W.; Dance, D.A.; Rattanavong, S.; et al. Three phylogenetic groups have driven the recent population expansion of Cryptococcus neoformans. Nat. Commun. 2019, 10, 2035. [Google Scholar] [CrossRef]
- Firacative, C.; Roe, C.C.; Malik, R.; Ferreira-Paim, K.; Escandón, P.; Sykes, J.E.; Castañón-Olivares, L.R.; Contreras-Peres, C.; Samayoa, B.; Sorrell, T.C.; et al. MLST and Whole-Genome-Based Population Analysis of Cryptococcus gattii VGIII Links Clinical, Veterinary and Environmental Strains, and Reveals Divergent Serotype Specific Sub-populations and Distant Ancestors. PLoS Negl. Trop. Dis. 2016, 10, e0004861. [Google Scholar] [CrossRef] [Green Version]
- Patino, L.H.; Castillo-Castaneda, A.; Munoz, M.; Muskus, C.; Rivero-Rodriguez, M.; Perez-Doria, A.; Bejarano, E.E.; Ramirez, J.D. Revisiting the heterogeneous global genomic population structure of Leishmania infantum. Microb. Genom. 2021, 7, 000640. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Chen, Y.; Farrer, R.A.; Giamberardino, C.; Sakthikumar, S.; Jones, A.; Yang, T.; Tenor, J.L.; Wagih, O.; Van Wyk, M.; Govender, N.P.; et al. Microevolution of Serial Clinical Isolates of Cryptococcus neoformans var. grubii and C. gattii. mBio 2017, 8, e00166-17. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Janbon, G.; Ormerod, K.L.; Paulet, D.; Byrnes, E.J., 3rd; Yadav, V.; Chatterjee, G.; Mullapudi, N.; Hon, C.C.; Billmyre, R.B.; Brunel, F.; et al. Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation. PLoS Genet. 2014, 10, e1004261. [Google Scholar] [CrossRef]
- Dumetz, F.; Imamura, H.; Sanders, M.; Seblova, V.; Myskova, J.; Pescher, P.; Vanaerschot, M.; Meehan, C.J.; Cuypers, B.; De Muylder, G.; et al. Modulation of Aneuploidy in Leishmania donovani during Adaptation to Different In Vitro and In Vivo Environments and Its Impact on Gene Expression. mBio 2017, 8, e00599-17. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Rodeghier, B.; Fan, J.J.; McClelland, C.M.; Wickes, B.L.; Chaturvedi, V. Direct PCR of Cryptococcus neoformans MAT alpha and MATa pheromones to determine mating type, ploidy, and variety: A tool for epidemiological and molecular pathogenesis studies. J. Clin. Microbiol. 2000, 38, 2007–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, W.; Castaneda, A.; Jackson, S.; Huynh, M.; Castaneda, E. IberoAmerican Cryptococcal Study G. Molecular typing of IberoAmerican Cryptococcus neoformans isolates. Emerg. Infect. Dis. 2003, 9, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Vanhove, M.; Beale, M.A.; Rhodes, J.; Chanda, D.; Lakhi, S.; Kwenda, G.; Molloy, S.; Karunaharan, N.; Stone, N.; Harrison, T.S.; et al. Genomic epidemiology of Cryptococcus yeasts identifies adaptation to environmental niches underpinning infection across an African HIV/AIDS cohort. Mol. Ecol. 2017, 26, 1991–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, J.; Desjardins, C.A.; Sykes, S.M.; Beale, M.A.; Vanhove, M.; Sakthikumar, S.; Chen, Y.; Gujja, S.; Saif, S.; Chowdhary, A.; et al. Tracing Genetic Exchange and Biogeography of Cryptococcus neoformans var. grubii at the Global Population Level. Genetics 2017, 207, 327–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira-Paim, K.; Andrade-Silva, L.; Fonseca, F.M.; Ferreira, T.B.; Mora, D.J.; Andrade-Silva, J.; Khan, A.; Dao, A.; Reis, E.C.; Almeida, M.T.; et al. MLST-Based Population Genetic Analysis in a Global Context Reveals Clonality amongst Cryptococcus neoformans var. grubii VNI Isolates from HIV Patients in Southeastern Brazil. PLoS Negl. Trop. Dis. 2017, 11, e0005223. [Google Scholar] [CrossRef] [Green Version]
- Farrer, R.A.; Borman, A.M.; Inkster, T.; Fisher, M.C.; Johnson, E.M.; Cuomo, C.A. Genomic epidemiology of a Cryptococcus neoformans case cluster in Glasgow, Scotland, 2018. Microb. Genom. 2021, 7, 000537. [Google Scholar] [CrossRef]
- Braunsdorf, C.; LeibundGut-Landmann, S. Modulation of the Fungal-Host Interaction by the Intra-Species Diversity of C. albicans. Pathogens 2018, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Sitterle, E.; Maufrais, C.; Sertour, N.; Palayret, M.; d’Enfert, C.; Bougnoux, M.E. Within-Host Genomic Diversity of Candida albicans in Healthy Carriers. Sci. Rep. 2019, 9, 2563. [Google Scholar] [CrossRef] [Green Version]
- Grohs Ferrareze, P.A.; Maufrais, C.; Silva Araujo Streit, R.; Priest, S.J.; Cuomo, C.A.; Heitman, J.; Staats, C.C.; Janbon, G. Application of an optimized annotation pipeline to the Cryptococcus deuterogattii genome reveals dynamic primary metabolic gene clusters and genomic impact of RNAi loss. G3 2021, 11, jkaa070. [Google Scholar] [CrossRef]
- Dunn, M.J.; Kinney, G.M.; Washington, P.M.; Berman, J.; Anderson, M.Z. Functional diversification accompanies gene family expansion of MED2 homologs in Candida albicans. PLoS Genet. 2018, 14, e1007326. [Google Scholar] [CrossRef]
- Ropars, J.; Maufrais, C.; Diogo, D.; Marcet-Houben, M.; Perin, A.; Sertour, N.; Mosca, K.; Permal, E.; Laval, G.; Bouchier, C.; et al. Gene flow contributes to diversification of the major fungal pathogen Candida albicans. Nat. Commun. 2018, 9, 2253. [Google Scholar] [CrossRef] [Green Version]
- Del Olmo Toledo, V.; Puccinelli, R.; Fordyce, P.M.; Perez, J.C. Diversification of DNA binding specificities enabled SREBP transcription regulators to expand the repertoire of cellular functions that they govern in fungi. PLoS Genet. 2018, 14, e1007884. [Google Scholar] [CrossRef]
- Kwon-Chung, K.J.; Chang, Y.C. Aneuploidy and drug resistance in pathogenic fungi. PLoS Pathog. 2012, 8, e1003022. [Google Scholar] [CrossRef]
- Chang, Y.C.; Khanal Lamichhane, A.; Kwon-Chung, K.J. Cryptococcus neoformans, Unlike Candida albicans, Forms Aneuploid Clones Directly from Uninucleated Cells under Fluconazole Stress. mBio 2018, 9, e01290-18. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.H.; Sephton-Clark, P.; Tenor, J.L.; Toffaletti, D.L.; Giamberardino, C.; Haverkamp, M.; Cuomo, C.A.; Perfect, J.R. Gene Expression of Diverse Cryptococcus Isolates during Infection of the Human Central Nervous System. mBio 2021, 12, e0231321. [Google Scholar] [CrossRef]
- Chayakulkeeree, M.; Sorrell, T.C.; Siafakas, A.R.; Wilson, C.F.; Pantarat, N.; Gerik, K.J.; Boadle, R.; Djordjevic, J.T. Role and mechanism of phosphatidylinositol-specific phospholipase C in survival and virulence of Cryptococcus neoformans. Mol. Microbiol. 2008, 69, 809–826. [Google Scholar]
- Olson, G.M.; Fox, D.S.; Wang, P.; Alspaugh, J.A.; Buchanan, K.L. Role of protein O-mannosyltransferase Pmt4 in the morphogenesis and virulence of Cryptococcus neoformans. Eukaryot Cell 2007, 6, 222–234. [Google Scholar] [CrossRef] [Green Version]
- Altamirano, S.; Li, Z.; Fu, M.S.; Ding, M.; Fulton, S.R.; Yoder, J.M.; Tran, V.; Nielsen, K. The Cyclin Cln1 Controls Polyploid Titan Cell Formation following a Stress-Induced G2 Arrest in Cryptococcus. mBio 2021, 12, e0250921. [Google Scholar] [CrossRef]
- Liu, O.W.; Chun, C.D.; Chow, E.D.; Chen, C.; Madhani, H.D.; Noble, S.M. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell 2008, 135, 174–188. [Google Scholar] [CrossRef] [Green Version]
- Goebels, C.; Thonn, A.; Gonzalez-Hilarion, S.; Rolland, O.; Moyrand, F.; Beilharz, T.H.; Janbon, G. Introns regulate gene expression in Cryptococcus neoformans in a Pab2p dependent pathway. PLoS Genet. 2013, 9, e1003686. [Google Scholar] [CrossRef] [Green Version]
- Selmecki, A.M.; Maruvka, Y.E.; Richmond, P.A.; Guillet, M.; Shoresh, N.; Sorenson, A.L.; De, S.; Kishony, R.; Michor, F.; Dowell, R.; et al. Polyploidy can drive rapid adaptation in yeast. Nature 2015, 519, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J.L.; Rokas, A. Copy Number Variation in Fungi and Its Implications for Wine Yeast Genetic Diversity and Adaptation. Front. Microbiol. 2018, 9, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stalder, L.; Oggenfuss, U.; Mohd-Assaad, N.; Croll, D. The population genetics of adaptation through copy number variation in a fungal plant pathogen. Mol. Ecol. 2022, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kirchhausen, T. Three ways to make a vesicle. Nat. Rev. Mol. Cell Biol. 2000, 1, 187–198. [Google Scholar] [CrossRef]
- Jung, W.H.; Sham, A.; Lian, T.; Singh, A.; Kosman, D.J.; Kronstad, J.W. Iron source preference and regulation of iron uptake in Cryptococcus neoformans. PLoS Pathog. 2008, 4, e45. [Google Scholar] [CrossRef] [Green Version]
- Hicks, J.K.; D’Souza, C.A.; Cox, G.M.; Heitman, J. Cyclic AMP-dependent protein kinase catalytic subunits have divergent roles in virulence factor production in two varieties of the fungal pathogen Cryptococcus neoformans. Eukaryot Cell 2004, 3, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.R.; Yang, L.; Sebetso, G.; Allen, R.; Doan, T.H.; Blundell, R.; Lui, E.Y.; Morrow, C.A.; Fraser, J.A. Characterization of the complete uric acid degradation pathway in the fungal pathogen Cryptococcus neoformans. PLoS ONE 2013, 8, e64292. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Ma, X.; Ma, L.; Zhen, S.; Na, Y.; Zhang, P.; Zhu, X. The RNA Helicase Ski2 in the Fungal Pathogen Cryptococcus neoformans Highlights Key Roles in Azoles Resistance and Stress Tolerance. Med. Mycol. 2022, 60, myac083. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate ID | Age | Sex | Department | AIDS | ST | RFLP | Cluster (WGS) |
|---|---|---|---|---|---|---|---|
| H0058-I-3589 | 44 | Male | Cundinamarca | Negative | 100 | VNII | Cluster-1 (VNII) |
| H0058-I-2583 | 37 | Male | Antioquia | Positive | 377 | VNI | Cluster-3 (VNIa) |
| H0058-I-2619 | 43 | Male | Antioquia | Positive | 377 | VNI | Cluster-3 (VNIa) |
| H0058-I-2642 | 48 | Female | Antioquia | Positive | 377 | VNI | Cluster-3 (VNIa) |
| H0058-I-2746 | 34 | Female | Valle | Positive | 5 | VNI | Cluster-3 (VNIa) |
| H0058-I-2881 | 68 | Female | Valle | Negative | 298 | VNI | Cluster-3 (VNIa) |
| H0058-I-3097 | 21 | Female | Antioquia | Positive | 95 | VNI | Cluster-3 (VNIa) |
| H0058-I-3102 | 31 | Male | Antioquia | Positive | 377 | VNI | Cluster-3 (VNIa) |
| H0058-I-3104 | 48 | Female | Meta | Positive | 5 | VNI | Cluster-3 (VNIa) |
| H0058-I-3463 | 31 | Male | Atlantico | Positive | 6 | VNI | Cluster-3 (VNIa) |
| H0058-I-1959 | 54 | Male | N. santander | Negative | 77 | VNI | Cluster-3 (VNIa) |
| H0058-I-2677 | 72 | Male | Antioquia | Positive | 95 | VNI | Cluster-3 (VNIa) |
| H0058-I-2713 | ND | ND | ND | ND | 95 | VNI | Cluster-3 (VNIa) |
| H0058-I-2926 | ND | ND | ND | ND | 95 | VNI | Cluster-3 (VNIa) |
| H0058-I-2503 | 33 | Male | N. Santander | Positive | 63 | VNI | Cluster-3 (VNIb-X) |
| H0058-I-2807 | 30 | Male | Antioquia | Positive | 23 | VNI | Cluster-3 (VNIb-X) |
| H0058-I-3746 | 48 | Male | Cauca | Negative | 2 | VNI | Cluster-3 (VNIb-X) |
| H0058-I-2641 | ND | ND | ND | ND | 2 | VNI | Cluster-3 (VNIb-X) |
| H0058-I-2672 | ND | ND | ND | ND | 2 | VNI | Cluster-3 (VNIb-X) |
| H0058-I-2720 | ND | ND | ND | ND | 2 | VNI | Cluster-3 (VNIb-X) |
| H0058-I-2257 | 26 | Male | Antioquia | Positive | 69 | VNI | Cluster-3 (VNIb-Y) |
| H0058-I-2527 | ND | ND | ND | ND | 69 | VNI | Cluster-3 (VNIb-Y) |
| H0058-I-2643 | 46 | Male | Antioquia | Positive | 69 | VNI | Cluster-3 (VNIb-Y) |
| H0058-I-2725 | 39 | Male | Antioquia | Positive | 69 | VNI | Cluster-3 (VNIb-Y) |
| H0058-I-2779 | 39 | Male | Antioquia | Positive | 69 | VNI | Cluster-3 (VNIb-Y) |
| H0058-I-2806 | 36 | Male | Antioquia | Positive | 69 | VNI | Cluster-3 (VNIb-Y) |
| H0058-I-2808 | 31 | Male | Antioquia | Positive | 69 | VNI | Cluster-3 (VNIb-Y) |
| H0058-I-2950 | 44 | Male | Valle | Positive | 69 | VNI | Cluster-3 (VNIb-Y) |
| H0058-I-3279 | ND | ND | ND | ND | 69 | VNI | Cluster-3 (VNIb-Y) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patiño, L.H.; Muñoz, M.; Ramírez, A.L.; Vélez, N.; Escandón, P.; Parra-Giraldo, C.-M.; Ramírez, J.D. A Landscape of the Genomic Structure of Cryptococcus neoformans in Colombian Isolates. J. Fungi 2023, 9, 135. https://doi.org/10.3390/jof9020135
Patiño LH, Muñoz M, Ramírez AL, Vélez N, Escandón P, Parra-Giraldo C-M, Ramírez JD. A Landscape of the Genomic Structure of Cryptococcus neoformans in Colombian Isolates. Journal of Fungi. 2023; 9(2):135. https://doi.org/10.3390/jof9020135
Chicago/Turabian StylePatiño, Luz Helena, Marina Muñoz, Angie Lorena Ramírez, Nórida Vélez, Patricia Escandón, Claudia-Marcela Parra-Giraldo, and Juan David Ramírez. 2023. "A Landscape of the Genomic Structure of Cryptococcus neoformans in Colombian Isolates" Journal of Fungi 9, no. 2: 135. https://doi.org/10.3390/jof9020135
APA StylePatiño, L. H., Muñoz, M., Ramírez, A. L., Vélez, N., Escandón, P., Parra-Giraldo, C.-M., & Ramírez, J. D. (2023). A Landscape of the Genomic Structure of Cryptococcus neoformans in Colombian Isolates. Journal of Fungi, 9(2), 135. https://doi.org/10.3390/jof9020135