
Characterization and Genome Analysis of Cladobotryum mycophilum, the Causal Agent of Cobweb Disease of Morchella sextelata in China

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Field Surveys
2.2. Isolation and Purification of the Fungal Pathogens
2.3. Morphological and Molecular Biological Characterization of Pathogens
2.3.1. Morphological Identification
2.3.2. Molecular Biology Identification
2.4. Pathogenicity Determination
2.5. Genome Sequencing and Assembly
2.6. Genome Component Prediction
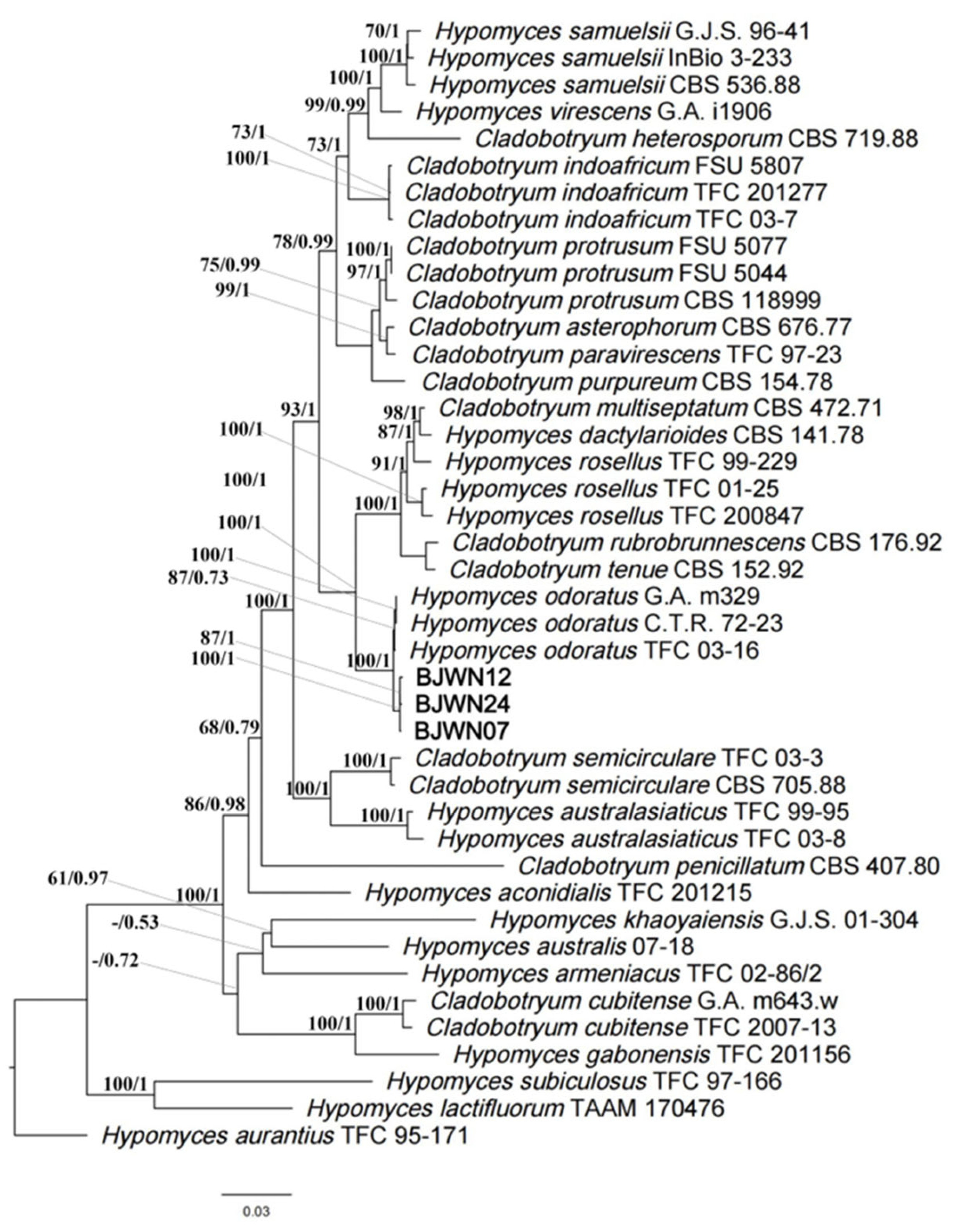
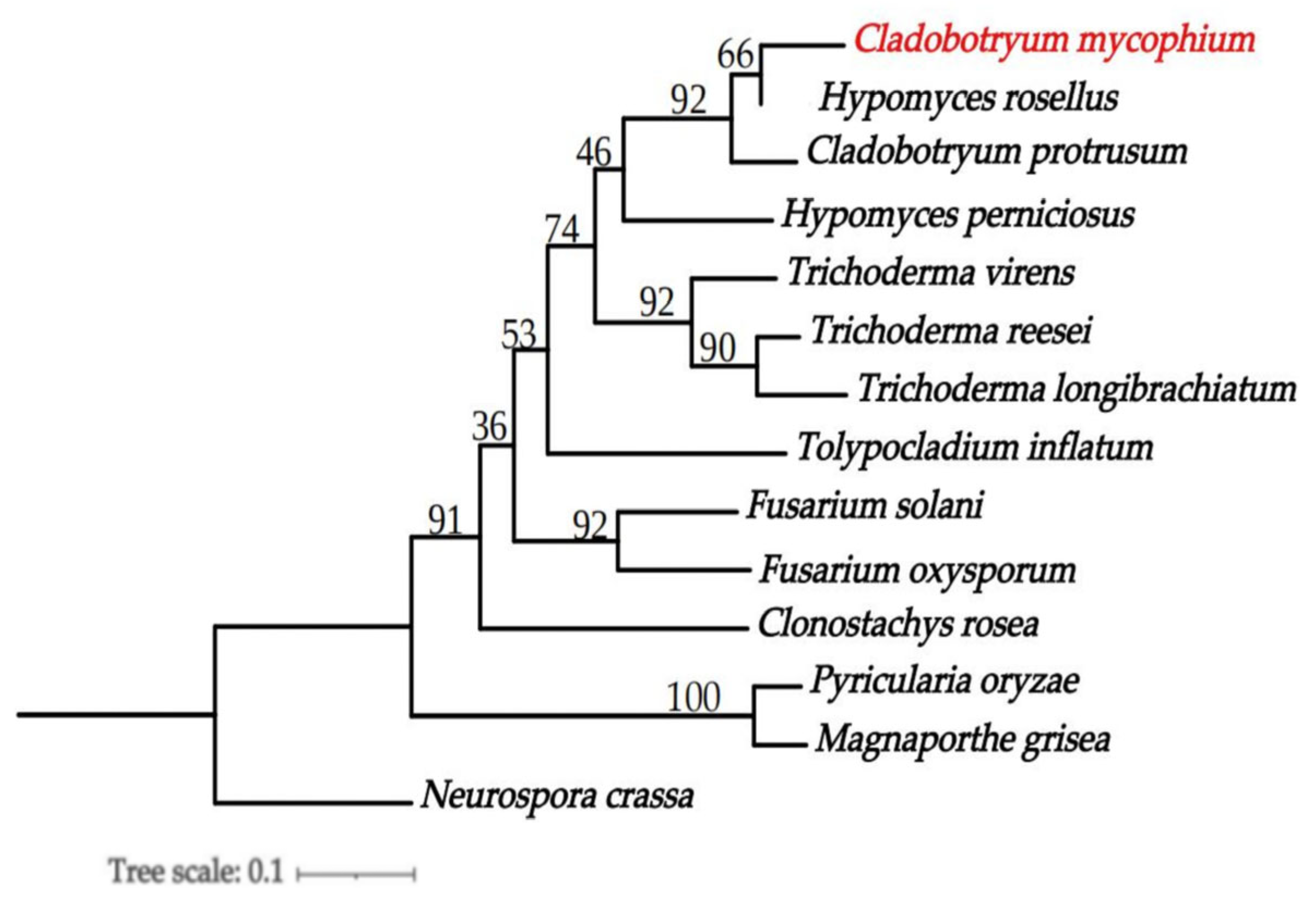
2.7. Phylogenomics Analysis of C. mycophilum
3. Results
3.1. Cobweb Disease Symptoms and Incidence
3.2. Identification of Pathogen
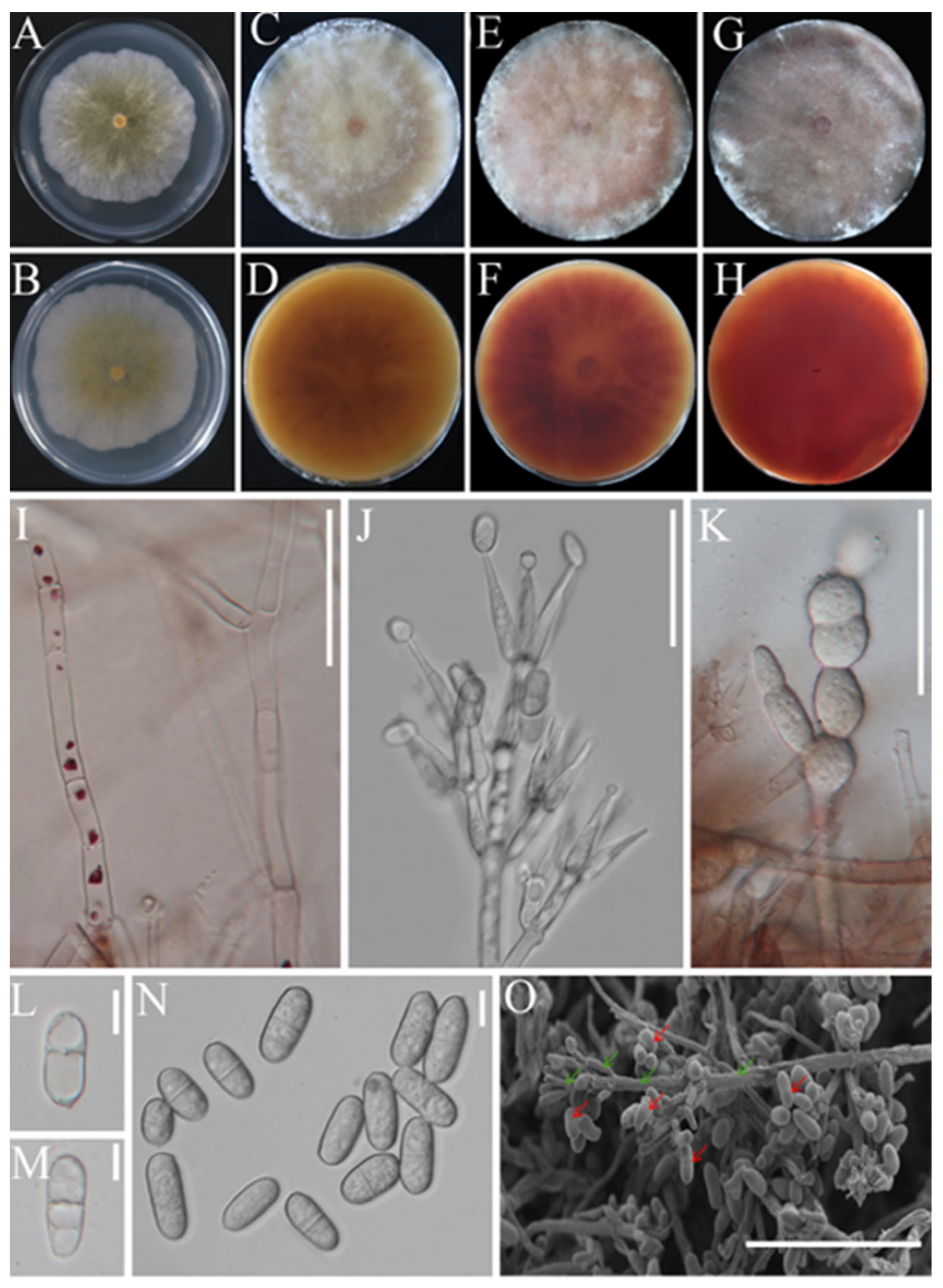
3.2.1. Morphological Characterization of the Pathogen
3.2.2. Molecular Identification of the Pathogen
3.3. Pathogenicity Determination
3.4. Genome Sequencing and Assembly
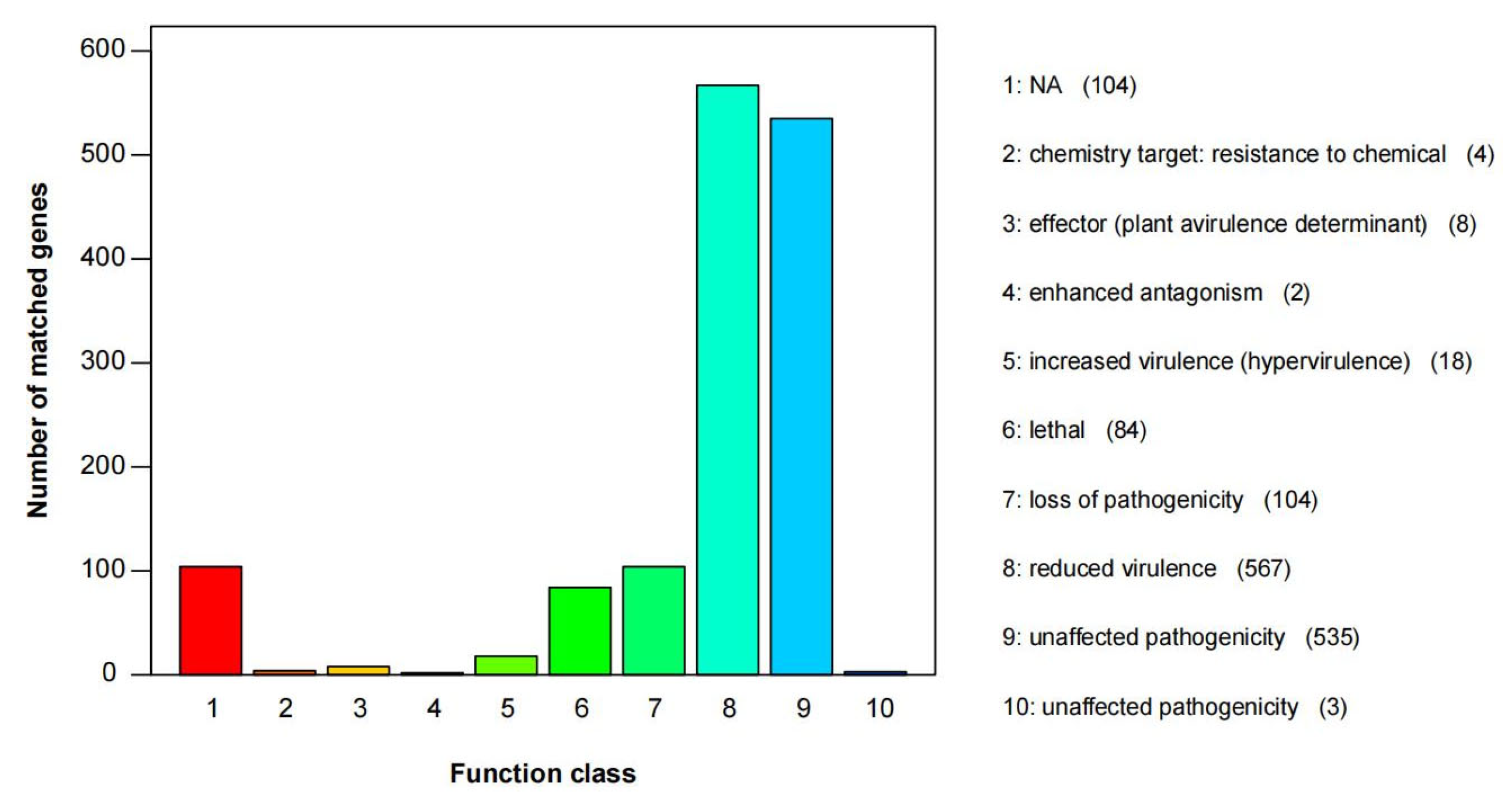
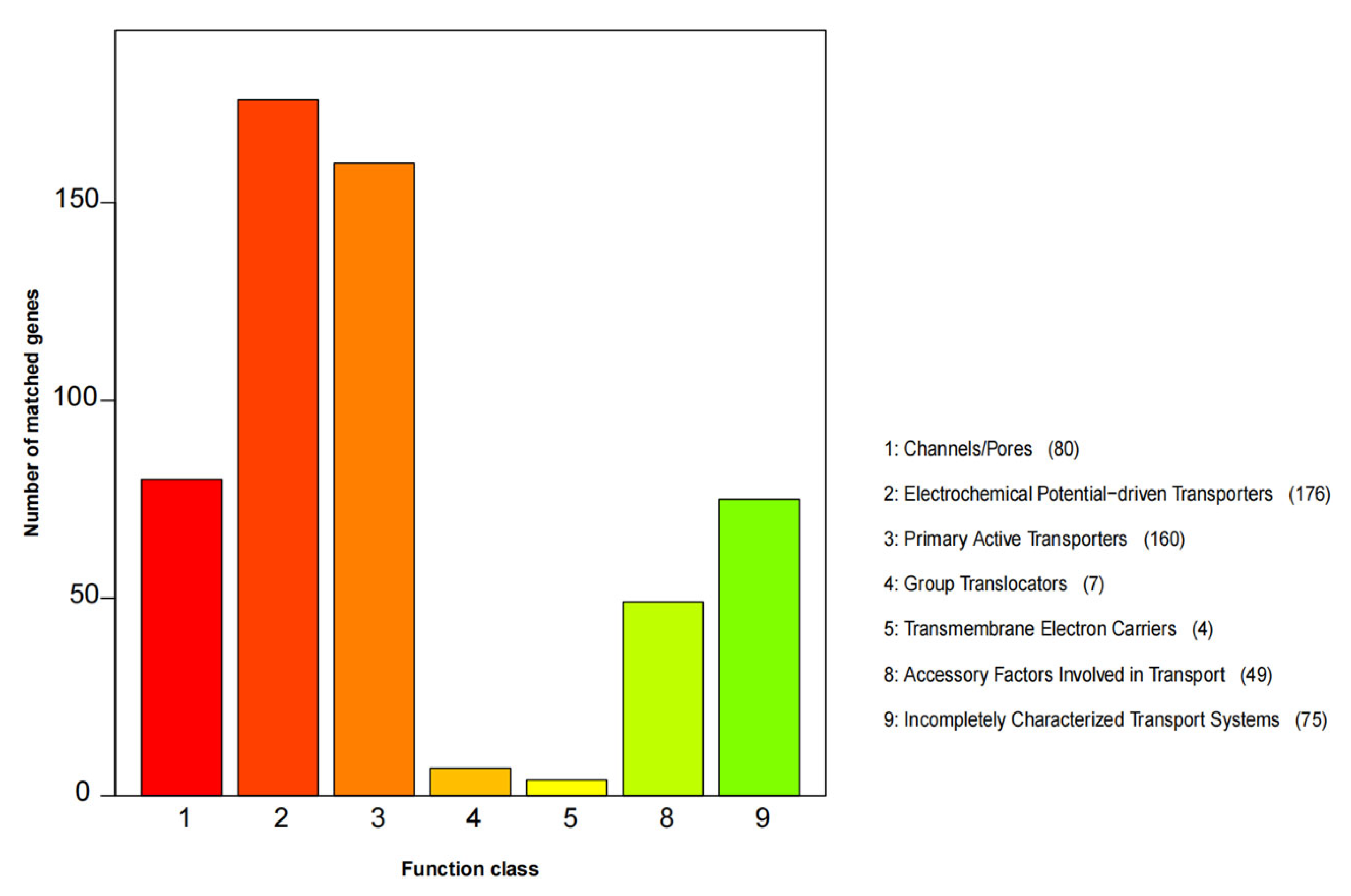
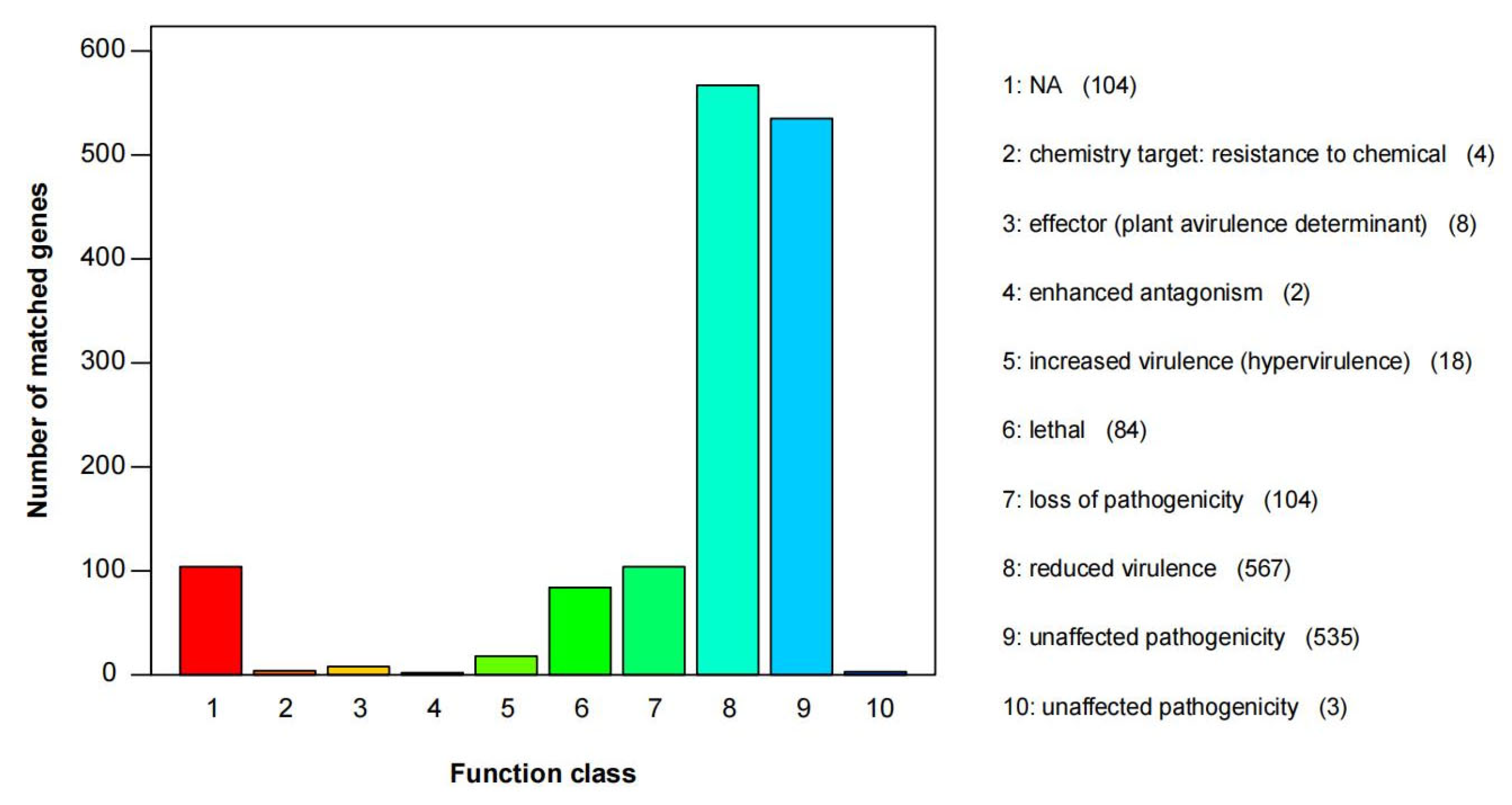
3.5. Gene Function Annotation
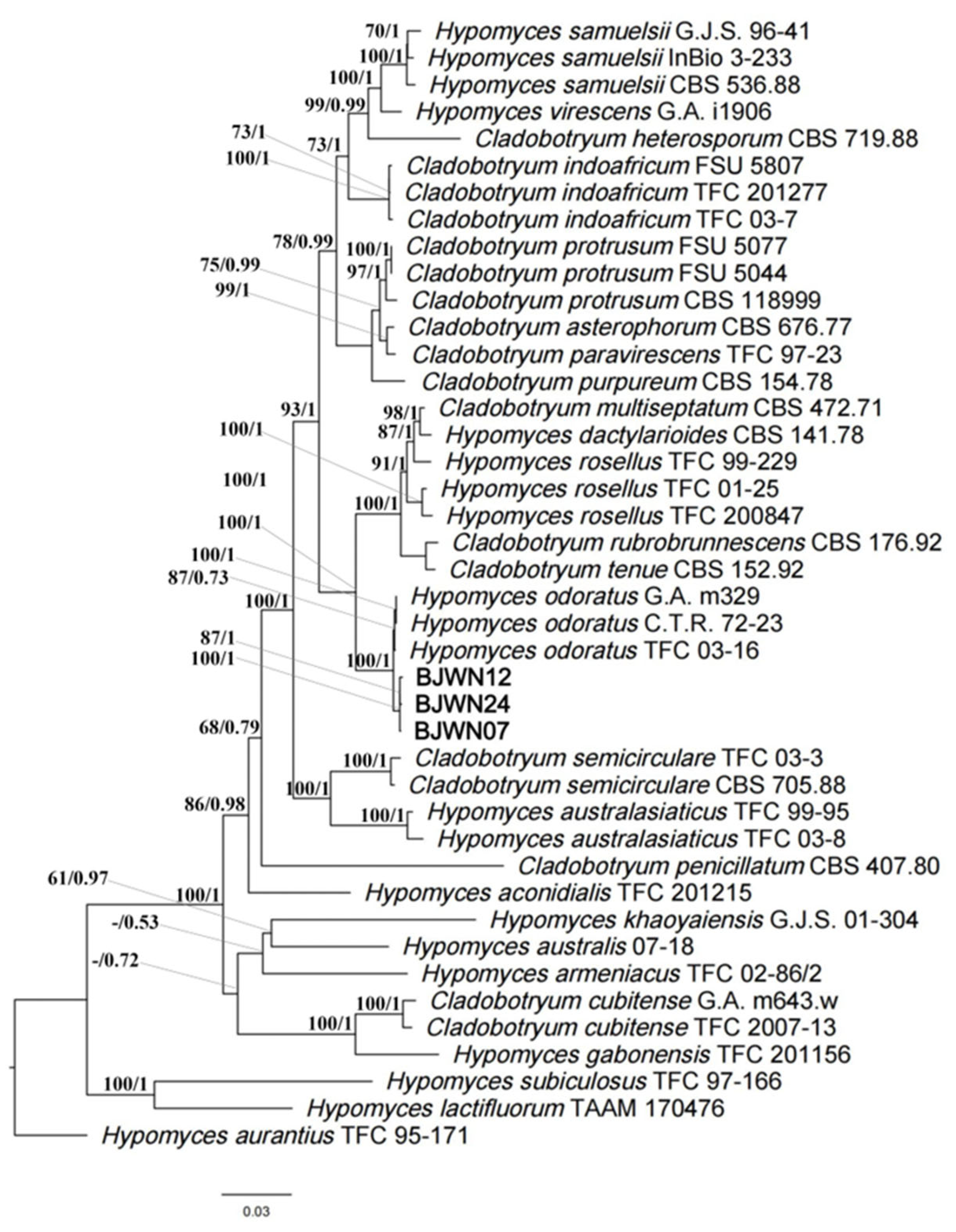
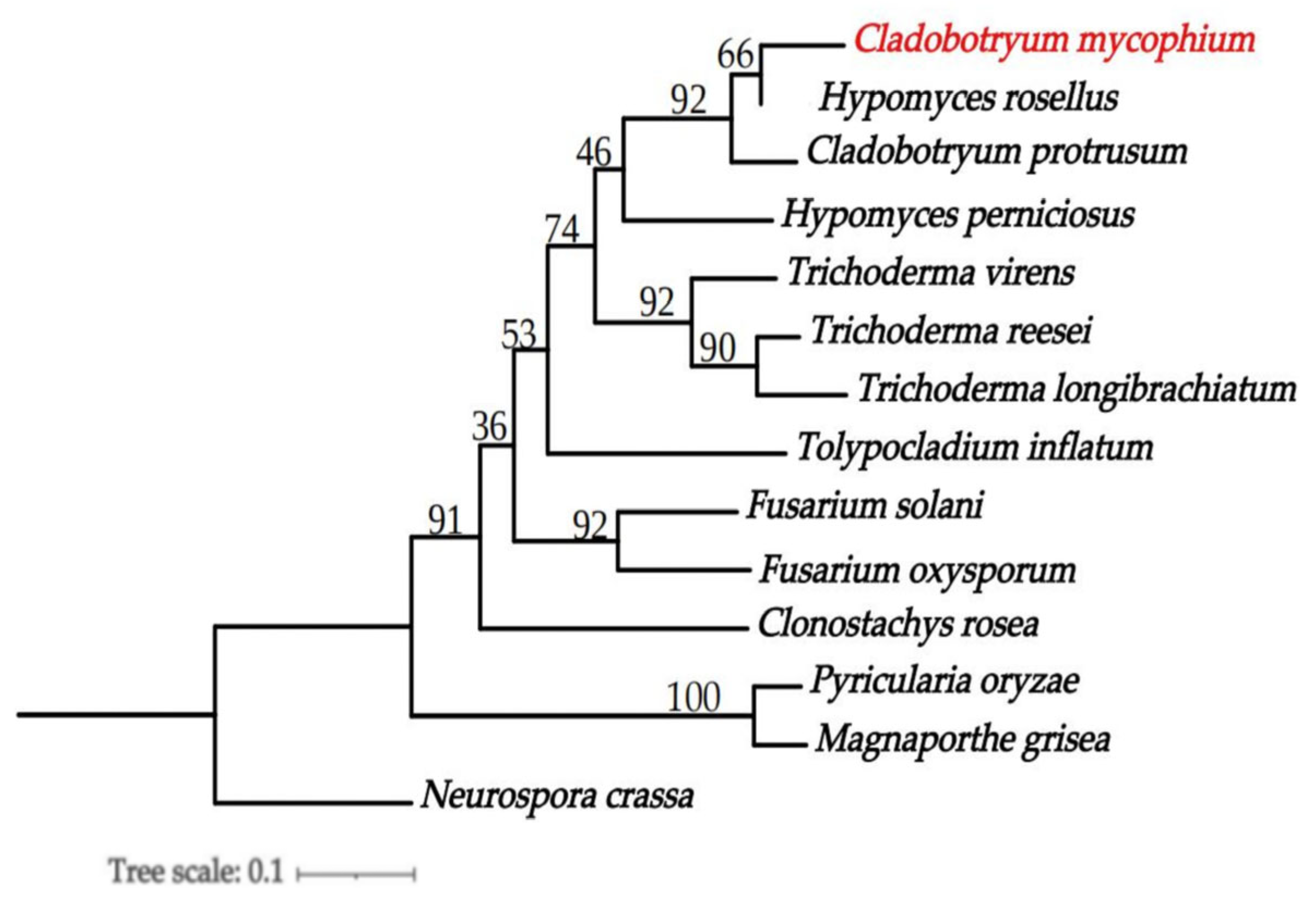
3.6. Phylogenomics Analysis of C. mycophilum
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.; Wu, C.; Sun, Y.; Li, T.; Fan, G. Cytoprotective Effect of Morchella esculenta Protein Hydrolysate and Its Derivative Against H2O2-Induced Oxidative Stress. Pol. J. Food Nutr. Sci. 2019, 69, 255–265. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, J.; Li, F.; Yang, Y.; Wu, S.; Ming, J. Antioxidant and Antiproliferative Activities of Modified Polysaccharides Originally Isolated from Morchella angusticepes Peck. J. Food Sci. 2019, 84, 448–456. [Google Scholar] [CrossRef]
- Liu, C.; Sun, Y.; Mao, Q.; Guo, X.; Li, P.; Liu, Y.; Xu, N. Characteristics and Antitumor Activity of Morchella esculenta Polysaccharide Extracted by Pulsed Electric Field. Int. J. Mol. Sci. 2016, 17, E986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wang, H.; Kang, Z.; Wu, Y.; Xing, Y.; Yang, Y. Antioxidant and Anti-Tumour Activity of Triterpenoid Compounds Isolated from Morchella Mycelium. Arch. Microbiol. 2020, 202, 1677–1685. [Google Scholar] [CrossRef]
- Cui, H.-L.; Chen, Y.; Wang, S.-S.; Kai, G.-Q.; Fang, Y.-M. Isolation, Partial Characterisation and Immunomodulatory Activities of Polysaccharide from Morchella esculenta: Properties of Polysaccharide from M. esculenta. J. Sci. Food Agric. 2011, 91, 2180–2185. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Peng, D.; Li, C.; Hu, X.; Bi, S.; Song, L.; Peng, B.; Zhu, J.; Chen, Y.; Yu, R. A New Polysaccharide Isolated from Morchella importuna Fruiting Bodies and Its Immunoregulatory Mechanism. Int. J. Biol. Macromol. 2019, 137, 8–19. [Google Scholar] [CrossRef]
- Zhao, Q.; Lv, M.; Li, L.; Huang, W.; Zhang, Y.; Hao, Z. Temptation and Trap of Morel Industry in China. J. Fungal Res. 2021, 19, 232–237. [Google Scholar] [CrossRef]
- Zhao, Q. Current Situation, Prospect and Suggestions of Morchella Industry in China. Edible Med. Mushrooms 2018, 26, 148–151. [Google Scholar]
- He, X.; Peng, W.; Miao, R.; Tang, J.; Chen, Y.; Liu, L.; Wang, D.; Gan, B. White Mold on Cultivated Morels Caused by Paecilomyces penicillatus. FEMS Microbio. Lett. 2017, 364, fnx037. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhou, J.; Wang, D.; He, X.; Tang, J.; Chen, Y.; Wang, J.; Peng, W. A New Stipe Rot Disease of the Cultivated Morchella sextelata. J. Fungal Res. 2021, 40, 2229–2243. [Google Scholar] [CrossRef]
- Lv, B.; Sun, Y.; Chen, Y.; Yu, H.; Mo, Q. First Report of Lecanicillium aphanocladii Causing Rot of Morchella sextelata in China. Plant Dis. 2022, 106, 3202. [Google Scholar] [CrossRef]
- Lan, Y.F.; Cong, Q.Q.; Wang, Q.W.; Tang, L.N.; Li, X.M.; Yu, Q.W.; Cui, X.; An, X.R.; Yu, C.X.; Kong, F.H.; et al. First Report of Cladobotryum protrusum Causing Cobweb Disease on Cultivated Morchella importuna. Plant Dis. 2020, 104, 977. [Google Scholar] [CrossRef]
- Fletcher, J.; Gaze, R. Mushroom Pest and Disease Control: A Colour Handbook, 1st ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 1–160. [Google Scholar] [CrossRef]
- Adie, B.; Grogan, H.; Archer, S.; Mills, P. Temporal and Spatial Dispersal of Cladobotryum Conidia in the Controlled Environment of a Mushroom Growing Room. Appl. Environ. Microbiol. 2006, 72, 7212–7217. [Google Scholar] [CrossRef] [Green Version]
- Carrasco, J.; Navarro, M.J.; Gea, F.J. Cobweb, A Serious Pathology in Mushroom Crops: A Review. Span. J. Agric. Res. 2017, 15, e10R01. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.Y.; Fu, Y.P.; Li, Y. Biological characteristics of Cladobotryum dendroides, a causal pathogen of Lentinula edodes cobweb disease. J. Fungal Res. 2019, 38, 646–657. [Google Scholar] [CrossRef]
- Potocnik, I.; Rekanovic, E.; Milijašević-Marčić, S.; Biljana, T.; Stepanovic, M. Morphological and Pathogenic Characteristics of the Fungus Cladobotryum dendroides, the Causal Agent of Cobweb Disease of the Cultivated Mushroom Agaricus Bisporus in Serbia. Pesticidi i Fitomedicina 2008, 23, 175–181. [Google Scholar] [CrossRef]
- Zhang, Q.-H.; Wang, W.; Li, C.-H.; Wen, Z.-Q. Biological characteristics of Hypomyces aurantius parasitic on Hypsizygus marmoreus. J. Fungal Res. 2015, 34, 350–356. [Google Scholar] [CrossRef]
- Kim, H.-K.; Seok, S.-J.; Kim, G.-P.; Moon, B.-J.; Terashita, T. Occurrence of Disease Caused by Cladobotryum varium on Flammulina velutipes in Korea. Kor. J. Mycol. 1999, 27, 415–419. [Google Scholar]
- Qin, W.T.; Li, J.; Zeng, Z.Q.; Wang, S.X.; Gao, L.; Rong, C.B.; Gao, Q.; Liu, Y. First Report of Cobweb Disease in Oudemansiella raphanipes Caused by Cladobotryum varium in Beijing, China. Plant Dis. 2021, 105, 4171. [Google Scholar] [CrossRef]
- Kirschner, R.; Arnold, G.; Chen, C.-J. Cladobotryum semicirculare sp. nov. (Hyphomycetes) from Commercially Grown Ganoderma tsugae in Taiwan and Other Basidiomycota in Cuba. Sydowia 2007, 59, 114–124. [Google Scholar]
- Wang, G.Z.; Ma, C.J.; Zhou, S.S.; Põldmaa, K.; Tamm, H.; Luo, Y.; Guo, M.P.; Ma, X.L.; Bian, Y.B.; Zhou, Y. First Report of Cobweb Disease of Auricularia polytricha Caused by Cladobotryum cubitense in Xuzhou, China. Plant Dis. 2018, 102, 1452. [Google Scholar] [CrossRef]
- Wang, G.Z.; Guo, M.P.; Bian, Y.B. First Report of Cladobotryum protrusum Causing Cobweb Disease on the Edible Mushroom Coprinus comatus. Plant Dis. 2015, 99, 287. [Google Scholar] [CrossRef] [PubMed]
- Chakwiya, A.; Van der Linde, E.J.; Chidamba, L.; Korsten, L. Diversity of Cladobotryum mycophilum Isolates Associated with Cobweb Disease of Agaricus bisporus in the South African Mushroom Industry. Eur. J. Plant Pathol. 2019, 154, 767–776. [Google Scholar] [CrossRef]
- Zuo, B.; Lu, B.; Liu, X.; Wang, Y.; Ma, G.; Wang, X.; Yang, L.; Liu, X.; Gao, J. First Report of Cladobotryum mycophilum Causing Cobweb on Ganoderma lucidum Cultivated in Jilin Province, China. Plant Dis. 2016, 100, 1239. [Google Scholar] [CrossRef]
- Kim, M.K.; Lee, Y.H.; Cho, K.M.; Lee, J.Y. First Report of Cobweb Disease Caused by Cladobotryum mycophilum on the Edible Mushroom Pleurotus eryngii in Korea. Plant Dis. 2012, 96, 1374. [Google Scholar] [CrossRef]
- Gea, F.J.; Navarro, M.J.; Suz, L.M. First Report of Cladobotryum mycophilum Causing Cobweb on Cultivated King Oyster Mushroom in Spain. Plant Dis. 2011, 95, 1030. [Google Scholar] [CrossRef]
- Sossah, F.L.; Liu, Z.; Yang, C.; Okorley, B.A.; Sun, L.; Fu, Y.; Li, Y. Genome Sequencing of Cladobotryum protrusum Provides Insights into the Evolution and Pathogenic Mechanisms of the Cobweb Disease Pathogen on Cultivated Mushroom. Genes 2019, 10, 124. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Liu, X.; Peng, B.; Liu, P.; Li, Z.; Dai, Y.; Xiao, S. Genomic Features of Cladobotryum dendroides, Which Causes Cobweb Disease in Edible Mushrooms, and Identification of Genes Related to Pathogenicity and Mycoparasitism. Pathogens 2020, 9, 232. [Google Scholar] [CrossRef] [Green Version]
- Seifert, K.A.; Gams, W. The Genera of Hyphomycetes—2011 Update. Persoonia 2011, 27, 119–129. [Google Scholar] [CrossRef] [Green Version]
- White; Bruns, T.; Lee, S.; Taylor, J. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols. A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar] [CrossRef]
- Carbone, I.; Kohn, L.M. A Method for Designing Primer Sets for Speciation Studies in Filamentous ascomycetes. Mycologia 1999, 91, 553–556. [Google Scholar] [CrossRef]
- Primers for Elongation Factor 1-α-AFTOL-Douding Web. Available online: https://www.docin.com/p-1613748809.html (accessed on 5 November 2022).
- Liu, Y.J.; Whelen, S.; Hall, B.D. Phylogenetic Relationships among Ascomycetes: Evidence from an RNA Polymerse II Subunit. Mol. Biol. Evol. 1999, 16, 1799–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T. Bioedit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/ NT. Nucl. Acids. Symp. Ser. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.J.; Lee, E.H.; Yoon, Y.; Chua, B.; Son, A. Portable Lysis Apparatus for Rapid Single-Step DNA Extraction of Bacillus subtilis. Proc. Soc. Appl. Bact. 2016, 120, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG Resource for Deciphering the Genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From Genomics to Chemical Genomics: New Developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded Microbial Genome Coverage and Improved Protein Family Annotation in the COG Database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating Some Redundancy Significantly Speeds up Clustering of Large Protein Databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Saier, M.H.; Reddy, V.S.; Tsu, B.V.; Ahmed, M.S.; Li, C.; Moreno-Hagelsieb, G. The Transporter Classification Database (TCDB): Recent Advances. Nucleic Acids Res. 2016, 44, D372–D379. [Google Scholar] [CrossRef] [Green Version]
- Bairoch, A.; Apweiler, R. The SWISS-PROT Protein Sequence Database and Its Supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating Signal Peptides from Transmembrane Regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. AntiSMASH: Rapid Identification, Annotation and Analysis of Secondary Metabolite Biosynthesis Gene Clusters in Bacterial and Fungal Genome Sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Urban, M.; Pant, R.; Raghunath, A.; Irvine, A.G.; Pedro, H.; Hammond-Kosack, K.E. The Pathogen-Host Interactions Database (PHI-Base): Additions and Future Developments. Nucleic Acids Res. 2015, 43, D645–D655. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Sossah, F.L.; Sun, L.; Fu, Y.; Li, Y. Genome Analysis of Hypomyces perniciosus, the Causal Agent of Wet Bubble Disease of Button Mushroom (Agaricus bisporus). Genes 2019, 10, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druzhinina, I.S.; Chenthamara, K.; Zhang, J.; Atanasova, L.; Yang, D.; Miao, Y.; Rahimi, M.J.; Grujic, M.; Cai, F.; Pourmehdi, S.; et al. Massive Lateral Transfer of Genes Encoding Plant Cell Wall-Degrading Enzymes to the Mycoparasitic Fungus Trichoderma from Its Plant-Associated Hosts. PLoS Genet. 2018, 14, e1007322. [Google Scholar] [CrossRef] [Green Version]
- Galagan, J.E.; Calvo, S.E.; Borkovich, K.A.; Selker, E.U.; Read, N.D.; Jaffe, D.; FitzHugh, W.; Ma, L.-J.; Smirnov, S.; Purcell, S.; et al. The Genome Sequence of the Filamentous Fungus Neurospora crassa. Nature 2003, 422, 859–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, R.A.; Talbot, N.J.; Ebbole, D.J.; Farman, M.L.; Mitchell, T.K.; Orbach, M.J.; Thon, M.; Kulkarni, R.; Xu, J.-R.; Pan, H.; et al. The Genome Sequence of the Rice Blast Fungus Magnaporthe grisea. Nature 2005, 434, 980–986. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Singh, P.K.; Gupta, D.K.; Mahato, A.K.; Sarkar, C.; Rathour, R.; Singh, N.K.; Sharma, T.R. Analysis of Magnaporthe oryzae Genome Reveals a Fungal Effector, Which Is Able to Induce Resistance Response in Transgenic Rice Line Containing Resistance Gene, Pi54. Front. Plant Sci. 2016, 7, 1140. [Google Scholar] [CrossRef] [Green Version]
- Warmington, R.J.; Kay, W.; Jeffries, A.; O’Neill, P.; Farbos, A.; Moore, K.; Bebber, D.P.; Studholme, D.J. High-Quality Draft Genome Sequence of the Causal Agent of the Current Panama Disease Epidemic. Microbiol. Resour. Announc. 2019, 8, e00904–e00919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushley, K.E.; Raja, R.; Jaiswal, P.; Cumbie, J.S.; Nonogaki, M.; Boyd, A.E.; Owensby, C.A.; Knaus, B.J.; Elser, J.; Miller, D.; et al. The Genome of Tolypocladium inflatum: Evolution, Organization, and Expression of the Cyclosporin Biosynthetic Gene Cluster. PLoS Genet. 2013, 9, e1003496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubicek, C.P.; Herrera-Estrella, A.; Seidl-Seiboth, V.; Martinez, D.A.; Druzhinina, I.S.; Thon, M.; Zeilinger, S.; Casas-Flores, S.; Horwitz, B.A.; Mukherjee, P.K.; et al. Comparative Genome Sequence Analysis Underscores Mycoparasitism as the Ancestral Life Style of Trichoderma. Genome Biol. 2011, 12, R40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, D.; Berka, R.M.; Henrissat, B.; Saloheimo, M.; Arvas, M.; Baker, S.E.; Chapman, J.; Chertkov, O.; Coutinho, P.M.; Cullen, D.; et al. Genome Sequencing and Analysis of the Biomass-Degrading Fungus Trichoderma reesei (Syn. Hypocrea jecorina). Nat. Biotechnol. 2008, 26, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, M.; Durling, M.B.; Choi, J.; Kosawang, C.; Lackner, G.; Tzelepis, G.D.; Nygren, K.; Dubey, M.K.; Kamou, N.; Levasseur, A.; et al. Insights on the Evolution of Mycoparasitism from the Genome of Clonostachys rosea. Genome Biol. Evol. 2015, 7, 465–480. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Back, C.-G.; Lee, C.-Y.; Seo, G.-S.; Jung, H.-Y. Characterization of Species of Cladobotryum Which Cause Cobweb Disease in Edible Mushrooms Grown in Korea. Mycobiology 2012, 40, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Gea, F.J.; Navarro, M.J.; Suz, L.M. First Report of Cobweb Disease Caused by Cladobotryum dendroides on Shiitake Mushroom (Lentinula edodes) in Spain. Plant Dis. 2018, 5, 1030. [Google Scholar] [CrossRef]
- Gea, F.J.; Carrasco, J.; Navarro, M. Characterization and Pathogenicity of Cladobotryum mycophilum in Spanish Pleurotus eryngii Mushroom Crops and its Sensitivity to Fungicides. Eur. J. Plant Pathol. 2016, 147, 189–194. [Google Scholar] [CrossRef]
- Wu, X.Y.; Li, Y. First Report of Cobweb Disease Caused by Cladobotryum mycophilum on Cultivated Shiitake Mushroom (Lentinula edodes) in China. Plant Dis. 2020, 104, 573. [Google Scholar] [CrossRef]
- Tian, F.H.; Li, C.T.; Li, Y. First Report of Cladobotryum varium Causing Cobweb Disease of Pleurotus eryngii var. tuoliensis in China. Plant Dis. 2018, 102, 826. [Google Scholar] [CrossRef]
- Gea, F.J.; Navarro, M.J.; Suz, L.M. Cobweb Disease on Oyster Culinary-Medicinal Mushroom (Pleurotus ostreatus) Caused by the Mycoparasite Cladobotryum mycophilum. J. Plant Pathol. 2019, 101, 349–354. [Google Scholar] [CrossRef]
- Carrasco, J.; Navarro, M.; Santos, M.; Diánez, F.; Gea, F.J. Incidence, Identification and Pathogenicity of Cladobotryum mycophilum, Causal Agent of Cobweb Disease on Agaricus bisporus Mushroom Crops in Spain. Ann. Appl. Biol. 2016, 168, 214–224. [Google Scholar] [CrossRef]
- Mohanta, T.K.; Bae, H. The diversity of fungal genome. Biol. Proced Online 2015, 17, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashyal, B.M.; Rawat, K.; Sharma, S.; Kulshreshtha, D.; Gopala Krishnan, S.; Singh, A.K.; Dubey, H.; Solanke, A.U.; Sharma, T.R.; Aggarwal, R. Whole Genome Sequencing of Fusarium fujikuroi Provides Insight into the Role of Secretory Proteins and Cell Wall Degrading Enzymes in Causing Bakanae Disease of Rice. Front Plant Sci. 2017, 8, 2013. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.-S.; Xie, X.-L.; Liang, G.; Gong, F.; Wang, Y.; Wei, X.-Q.; Wang, Q.; Ji, Z.-L.; Chen, Q.-X. The GH18 Family of Chitinases: Their Domain Architectures, Functions and Evolutions. Glycobiology 2012, 22, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidl, V. Chitinases of Filamentous Fungi: A Large Group of Diverse Proteins with Multiple Physiological Functions. Fungal Biol. Rev. 2008, 22, 36–42. [Google Scholar] [CrossRef]
- Frandsen, R.J.N.; Nielsen, N.J.; Maolanon, N.; Sørensen, J.C.; Olsson, S.; Nielsen, J.; Giese, H. The Biosynthetic Pathway for Aurofusarin in Fusarium graminearum Reveals a Close Link between the Naphthoquinones and Naphthopyrones. Mol. Microbiol. 2006, 61, 1069–1080. [Google Scholar] [CrossRef]
- De Wit, P.J.G.M.; Mehrabi, R.; Van den Burg, H.A.; Stergiopoulos, I. Fungal Effector Proteins: Past, Present and Future. Mol. Plant Pathol. 2009, 10, 735–747. [Google Scholar] [CrossRef]
- Sperschneider, J.; Gardiner, D.M.; Dodds, P.N.; Tini, F.; Covarelli, L.; Singh, K.B.; Manners, J.M.; Taylor, J.M. EffectorP: Predicting Fungal Effector Proteins from Secretomes Using Machine Learning. New Phytol. 2016, 210, 743–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Genbank Accession Numbers | ||
|---|---|---|---|---|
| ITS | TEF | RPB2 | ||
| C. asterophorum | CBS 676.77 | FN859395 | FN868712 | FN868649 |
| C. cubitense | G.A. m643.w | FN859397 | FN868714 | FN868651 |
| TFC 2007-13 | AM779857 | FN868715 | FN868652 | |
| C. heterosporum | CBS 719.88 | FN859398 | FN868716 | FN868653 |
| C. indoafrum | FSU 5807 | FN859399 | FN868717 | FN868654 |
| TFC 03-7 | FN859400 | FN868718 | FN868655 | |
| TFC 201277 | FN859401 | FN868719 | FN868656 | |
| C. multiseptatum | CBS 472.71 | FN859405 | FN868723 | FN868659 |
| C. mycophilum | BJWN07 | OP714368 | OP759638 | OP718561 |
| BJWN12 | OP714369 | OP759639 | OP718562 | |
| BJWN24 | OP714393 | OP759640 | OP718563 | |
| C. paravirescens | TFC 97-23 | FN859406 | FN868724 | FN868660 |
| C. penicillatum | CBS 407.80 | FN859407 | FN868725 | FN868661 |
| C. protrusum | CBS 118999 | FN859408 | FN868726 | FN868662 |
| FSU 5044 | FN859409 | FN868727 | FN868663 | |
| FSU 5077 | FN859410 | FN868728 | FN868664 | |
| C. purpureum | CBS 154.78 | FN859415 | FN868733 | FN868669 |
| C. rubrobrunnescens | CBS 176.92 | FN859416 | FN868734 | FN868670 |
| C. semicirculare | CBS 705.88 | FN859417 | FN868735 | FN868671 |
| TFC 03-3 | FN859418 | FN868736 | FN868672 | |
| C. tenue | CBS 152.92 | FN859420 | FN868738 | FN868674 |
| H. aconidialis | TFC 201215 | FN859455 | FN868774 | FN868710 |
| H. armeniacus | TFC 02-86/2 | FN859424 | FN868742 | FN868678 |
| H. aurantius | TFC 95-171 | FN859425 | FN868743 | FN868679 |
| H. australasiaticus | TFC 99-95 | FN859427 | FN868745 | FN868680 |
| TFC 03-8 | FN859428 | FN868746 | FN868681 | |
| H. australis | TFC 07-18 | AM779860 | FN868747 | FN868682 |
| H. dactylarioides | CBS 141.78 | FN859429 | FN868748 | FN868683 |
| H. gabonensis | TFC 201156 | FN859430 | FN868749 | FN868684 |
| H. khaoyaiensis | G.J.S. 01-304 | FN859431 | FN868750 | FN868685 |
| H. lactifluorum | TAAM 170476 | FN859432 | FN868751 | EU710773 |
| H. odoratus | C.T.R. 72-23 | FN859433 | FN868752 | FN868687 |
| G.A. m329 | FN859434 | FN868753 | FN868688 | |
| TFC 03-16 | FN859437 | FN868756 | FN868691 | |
| H. rosellus | TFC 99-229 | FN859441 | FN868759 | FN868695 |
| TFC 01-25 | FN859442 | FN868760 | FN868696 | |
| TFC 200847 | FN859438 | FN868761 | FN868692 | |
| H. samuelsii | CBS 536.88 | FN859444 | FN868763 | FN868698 |
| G.J.S. 96-41 | FN859448 | FN868766 | FN868702 | |
| InBio 3-233 | FN859450 | FN868768 | FN868704 | |
| H. subiculosus | TFC 97-166 | FN859452 | FN868770 | EU710776 |
| H. virescens | G.A. i1906 | FN859454 | FN868772 | FN868708 |
| Terms | BJWN07 |
|---|---|
| The number of reads | 300,757 |
| Data size (bp) | 5,272,956,422 |
| Minimum sequencing read length (bp) | 78 |
| N50 Contig Length (bp) | 18,317 |
| Maximum sequencing read length | 49,868 |
| Genome size (Mb) | 38.56 |
| Number of contigs | 10 |
| GC content | 47.84% |
| Coverage | 130× |
| Number of coding genes | 8428 |
| The number of RNAs | 330 |
| Database Used for Gene/Protein Annotation | Number of Genes |
|---|---|
| Nr | 7766 |
| GO | 5683 |
| KEGG | 7447 |
| KOG | 1964 |
| Pfam | 5683 |
| SwissProt | 3049 |
| TCDB | 551 |
| CAZy | 499 |
| Secretory_Protein | 661 |
| P450 | 155 |
| PHI | 1429 |
| DFVF | 443 |
| Classification | Number |
|---|---|
| Carbohydrate-binding molecule (CBM) | 50 |
| Carbohydrate Esterase (CE) | 28 |
| Glycoside hydrolases (GHs) | 249 |
| Glycosyltransferases (GTs) | 111 |
| Polysaccharide lyases (PLs) | 9 |
| Auxiliary activities (AA) | 52 |
| Total | 499 |
| Clusters | Clusters_Number | Gene_Number |
|---|---|---|
| T1PKS | 23 | 229 |
| siderophore | 1 | 2 |
| NRPS | 18 | 175 |
| T1PKS, terpene | 2 | 25 |
| NRPS-like, T1PKS | 2 | 21 |
| NRPS, NRPS-like, T1PKS | 4 | 85 |
| NRPS, T1PKS | 8 | 79 |
| NRPS, NRPS-like, T1PKS, indole, terpene | 1 | 29 |
| NRPS-like | 7 | 69 |
| terpene | 12 | 59 |
| Total | 78 | 773 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Cong, Y.; Sossah, F.L.; Lu, Y.; Kang, J.; Li, Y. Characterization and Genome Analysis of Cladobotryum mycophilum, the Causal Agent of Cobweb Disease of Morchella sextelata in China. J. Fungi 2023, 9, 411. https://doi.org/10.3390/jof9040411
Liu Z, Cong Y, Sossah FL, Lu Y, Kang J, Li Y. Characterization and Genome Analysis of Cladobotryum mycophilum, the Causal Agent of Cobweb Disease of Morchella sextelata in China. Journal of Fungi. 2023; 9(4):411. https://doi.org/10.3390/jof9040411
Chicago/Turabian StyleLiu, Zhenghui, Yunlong Cong, Frederick Leo Sossah, Yongzhong Lu, Jichuan Kang, and Yu Li. 2023. "Characterization and Genome Analysis of Cladobotryum mycophilum, the Causal Agent of Cobweb Disease of Morchella sextelata in China" Journal of Fungi 9, no. 4: 411. https://doi.org/10.3390/jof9040411
APA StyleLiu, Z., Cong, Y., Sossah, F. L., Lu, Y., Kang, J., & Li, Y. (2023). Characterization and Genome Analysis of Cladobotryum mycophilum, the Causal Agent of Cobweb Disease of Morchella sextelata in China. Journal of Fungi, 9(4), 411. https://doi.org/10.3390/jof9040411