
Genetic and Other Determinants for the Severity of Coccidioidomycosis: A Clinician’s Perspective
 , ,
, ,  and
and
Abstract
:1. Introduction
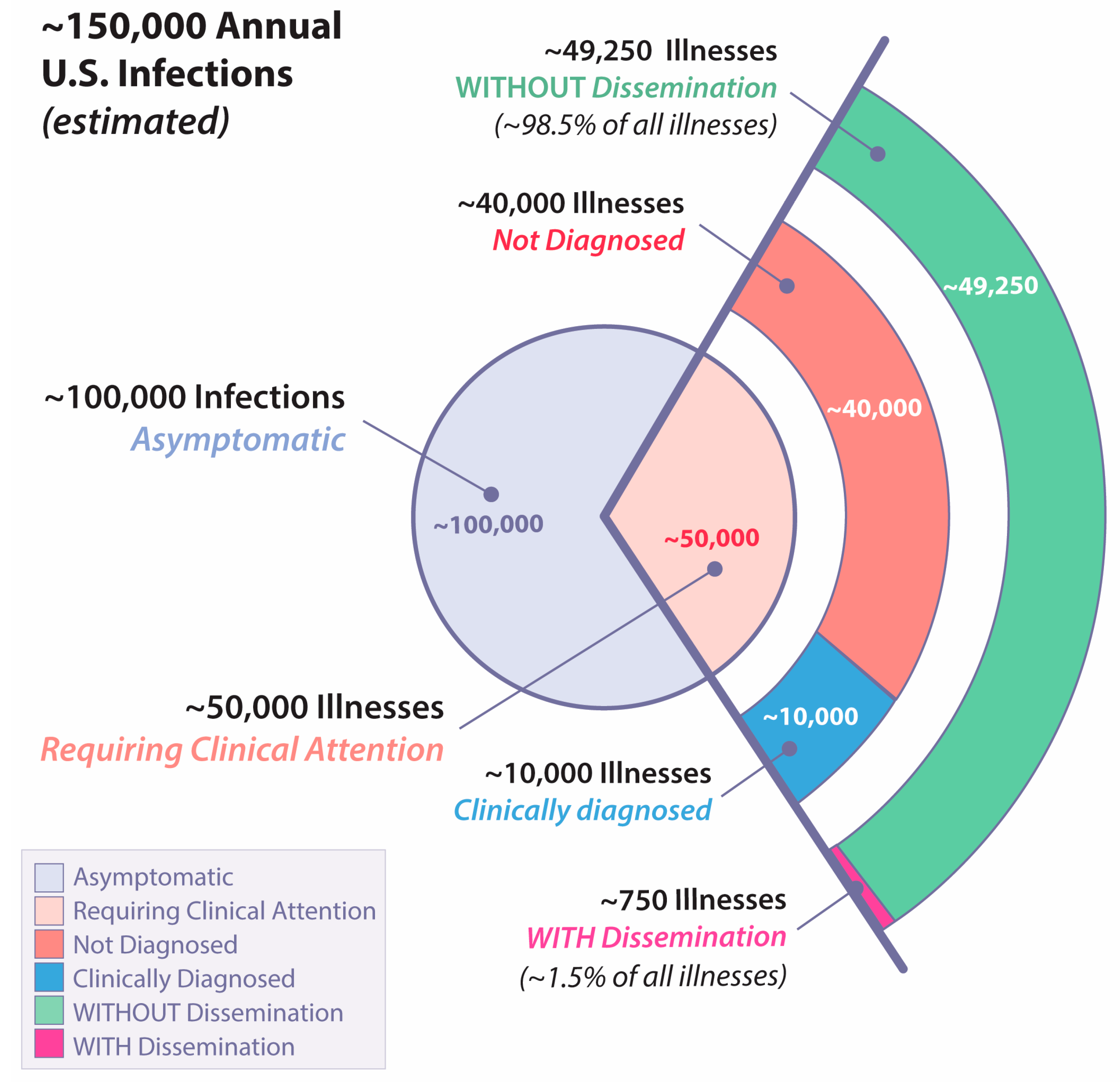
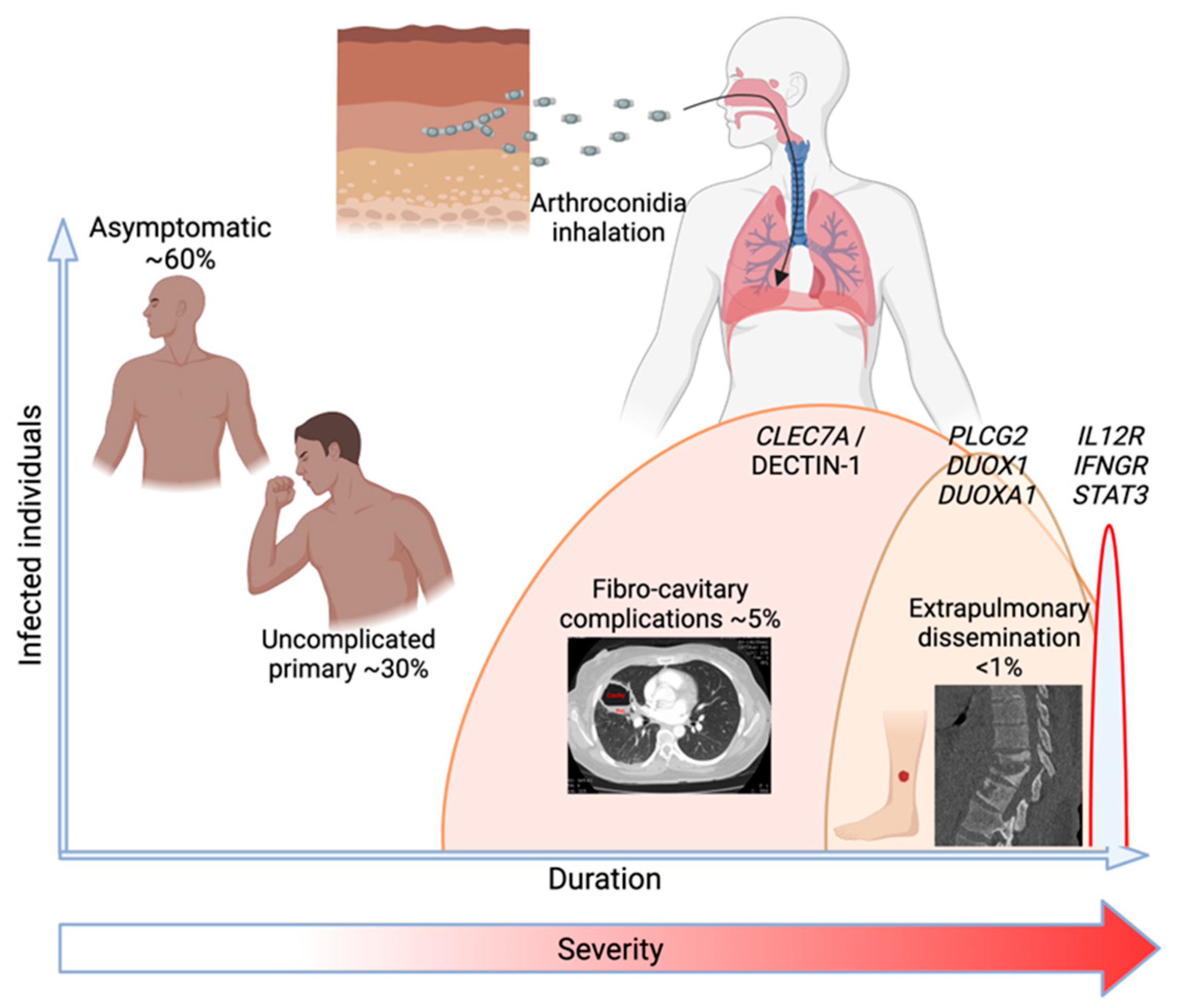
2. The Spectrum of Coccidioidomycosis in Humans
2.1. Asymptomatic Infection
2.2. Uncomplicated Primary Infection
2.3. Fibro-Cavitary Pulmonary Complications
2.4. Extrathoracic Dissemination
3. What Is Known about Genetic Determinants of Susceptibility to Coccidioidomycosis
4. Applying Current Knowledge Forward to a Unifying Model
5. Clinical Characteristics of Human Coccidioidomycosis Linked to Innate Signaling Defects
5.1. Disseminated Disease very Rarely Occurs in Multiple Generations
5.2. Clinically Significant Disseminated Disease Occurs as a Progression of the Initial Infection
5.3. Patients Who Develop Disseminated Coccidioidomycosis Usually Have an Absent or Self-Resolving Pulmonary Illness
5.4. Patients with Disseminated Infection Rarely if Ever Develop Pulmonary Illness from a Subsequent Respiratory Exposure to Coccidioides
5.5. Disseminated Disease can Reactivate as a Result of Profound Cellular Immunodeficiency
5.6. While Biologic Response Modifiers Increase the Risk of Disseminated Coccidioidal Infection, Many Patients Treated with Biologic Response Modifiers Control Their Infection without Complications
5.7. Disseminated Infections That Recur following Discontinuation of Antifungal Therapy Usually Do so at a Previously Identified Site of Infection
5.8. Non-Destructive Disseminated Infections Which Are Discovered Incidentally and Require no Medical Treatment also Occur
5.9. Patients with Disseminated Infection Occasionally Develop Dermal Hypersensitivity to Coccidioidal Antigens
6. Other Factors That Might Modify the Genetics Model
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dickson, E.C.; Gifford, M.A. Coccidioides infection (Coccidioidomycosis). II. The primary type of infection. Arch. Inern. Med. 1938, 62, 853–871. [Google Scholar]
- National Academies of Science, Engineering and Medicine. Impact and Control of Valley Fever––A Workshop; National Academies of Science, Engineering and Medicine: Irvine, CA, USA, 2022; Available online: https://www.nationalacademies.org/event/11-17-2022/impact-and-control-of-valley-fever-a-workshop (accessed on 14 March 2023).
- Mosberge, D. Dangerous Fungi Are Spreading Across U.S. as Temperatures Rise. Wall Str. J. 1 February 2023.
- Weaver, E.; Kolivras, K.N.; Thomas, R.Q.; Thomas, V.A.; Abbas, K.M. Environmental factors affecting ecological niche of Coccidioides species and spatial dynamics of valley fever in the United States. Spat. Spatiotemporal Epidemiol. 2020, 32, 100317. [Google Scholar] [CrossRef]
- Diaz, J.H. Travel-related risk factors for coccidioidomycosis. J. Travel Med. 2018, 25, tay027. [Google Scholar] [CrossRef]
- Mead, H.L.; Kollath, D.R.; Teixeira, M.M.; Roe, C.C.; Plude, C.; Nandurkar, N.; Donohoo, C.; O’Connor, B.L.W.; Terriquez, J.; Keim, P.; et al. Coccidioidomycosis in Northern Arizona: An Investigation of the Host, Pathogen, and Environment Using a Disease Triangle Approach. mSphere 2022, 7, e0035222. [Google Scholar] [CrossRef] [PubMed]
- Pappagianis, D.; Einstein, H. Tempest from Tehachapi takes toll or coccidioides conveyed aloft and afar. West. J. Med. 1978, 129, 527–530. [Google Scholar]
- Freedman, M.; Anderson, S.; Benedict, K.; McCotter, O.; Derado, G.; Hoekstra, R.M.; Galgiani, J.N.; Thompson, G.R., III; Rutherford, G.; Sunenshine, R. Accessed at Coccidioidomycosis Study Group 2022. Available online: http://coccistudygroup.com/wp-content/uploads/2017/11/CSG-61st-Annual-Proceedings-11-27-17.pdf (accessed on 30 June 2022).
- Pu, J.; Miranda, V.; Minior, D.; Reynolds, S.; Rayhorn, B.; Ellingson, K.D.; Galgiani, J.N. Improving Early Recognition of Coccidioidomycosis in Urgent Care Clinics: Analysis of an Implemented Education Program. Open Forum Infect. Dis. 2023, 10, ofac654. [Google Scholar] [CrossRef]
- Blair, J.E.; Ampel, N.M.; Hoover, S.E. Coccidioidomycosis in selected immunosuppressed hosts. Med. Mycol. 2019, 57 (Suppl. 1), S56–S63. [Google Scholar] [CrossRef]
- Huff, D.; Ampel, N.M.; Blair, J.E. Coccidioidomycosis and COVID-19 Infection. An Analysis from a Single Medical Center Within the Coccidioidal Endemic Area. Mycopathologia 2022, 187, 199–204. [Google Scholar] [CrossRef]
- Bercovitch, R.S.; Catanzaro, A.; Schwartz, B.S.; Pappagianis, D.; Watts, D.H.; Ampel, N.M. Coccidioidomycosis during pregnancy: A review and recommendations for management. Clin. Infect Dis. 2011, 53, 363–368. [Google Scholar] [CrossRef]
- Hsu, A.P.; Davis, J.; Chaput, A.L.; Powell, D.A.; Pouladi, N.; Lussier, Y.; Fierer, J.; Frelinger, J.A.; Galgiani, J.N.; Lionakis, M.; et al. Common Population Variants Cause Susceptibility to Disseminated Coccidioidomycosis. Open Forum Infect. Dis. 2020, 7 (Suppl. 1), S22–S23. [Google Scholar] [CrossRef]
- Werner, S.B.; Pappagianis, D.; Heindl, I.; Mickel, A. An epidemic of coccidioidomycosis among archeology students in northern California. N. Engl. J. Med. 1972, 286, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Shubitz, L.; Simons, J.; Perrill, R.; Orsborn, K.I.; Galgiani, J.N. Localization within a proline-rich antigen (Ag2/PRA) of protective antigenicity against infection with Coccidioides immitis in mice. Infect. Immun. 2002, 70, 3330–3335. [Google Scholar] [CrossRef] [PubMed]
- Jude, C.M.; Nayak, N.B.; Patel, M.K.; Deshmukh, M.; Batra, P. Pulmonary coccidioidomycosis: Pictorial review of chest radiographic and CT findings. Radiographics 2014, 34, 912–925. [Google Scholar] [CrossRef]
- Batra, P.; Batra, R.S. Thoracic coccidioidomycosis. Semin. Roentgenol. 1996, 31, 28–44. [Google Scholar] [CrossRef]
- Shemuel, J.; Bays, D.J.; Thompson, G.R., III; Reef, S.; Snyder, L.; Freifeld, A.J.; Huppert, M.; Salkin, D.; Wilson, M.D.; Galgiani, J.N. Natural history of pulmonary coccidioidomycosis: Further examination of the VA-Armed Forces Database. Med. Mycol. 2022, 60, myac054. [Google Scholar] [CrossRef]
- de Perio, M.A.; Materna, B.L.; Sondermeyer Cooksey, G.L.; Vugia, D.J.; Su, C.P.; Luckhaupt, S.E.; McNary, J.; Wilken, J.A. Occupational coccidioidomycosis surveillance and recent outbreaks in California. Med. Mycol. 2019, 57 (Suppl. 1), S41–S45. [Google Scholar] [CrossRef]
- Foley, C.G.T.; Christ, C.; Anderson, S.M. Impact of disseminated coccidioidomycosis in Arizona, 2007–2008. In Proceedings of the 55th Annual Coccidioidomycosis Study Group, University of California at Davis, Davis, CA, USA, 2 April 2011. [Google Scholar]
- Smith, C.E.; Beard, R.R.; Whiting, E.G.; Rosenberger, H.G. Varieties of coccidioidal infection in relation to the epidemiology and control of the disease. Am. J. Public Health 1946, 36, 1394–1402. [Google Scholar] [CrossRef]
- Kerrick, S.S.; Lundergan, L.L.; Galgiani, J.N. Coccidioidomycosis at a university health service. Am. Rev. Respir. Dis. 1985, 131, 100–102. [Google Scholar]
- Yozwiak, M.L.; Lundergan, L.L.; Kerrick, S.S.; Galgiani, J.N. Symptoms and routine laboratory abnormalities associated with coccidioidomycosis. West. J. Med. 1988, 149, 419–421. [Google Scholar]
- Valdivia, L.; Nix, D.; Wright, M.; Lindberg, E.; Fagan, T.; Lieberman, D.; Stoffer, T.; Ampel, N.M.; Galgiani, J.N. Coccidioidomycosis as a common cause of community-acquired pneumonia. Emerg. Infect. Dis. 2006, 12, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.M.; Blair, J.E.; Carey, E.J.; Wu, Q.; Smilack, J.D. Coccidioidal pneumonia, Phoenix, Arizona, USA, 2000–2004. Emerg. Infect. Dis. 2009, 15, 397–401. [Google Scholar]
- Galgiani, J.N.; Ampel, N.M.; Blair, J.E.; Catanzaro, A.; Geertsma, F.; Hoover, S.E.; Johnson, R.H.; Kusne, S.; Lisse, J.; MacDonald, J.D.; et al. 2016 Infectious Diseases Society of America (IDSA) Clinical Practice Guideline for the Treatment of Coccidioidomycosis. Clin. Infect. Dis. 2016, 63, e112–e146. [Google Scholar] [CrossRef] [PubMed]
- Ampel, N.M.; Giblin, A.; Mourani, J.P.; Galgiani, J.N. Factors and outcomes associated with the decision to treat primary pulmonary coccidioidomycosis. Clin. Infect. Dis. 2009, 48, 172–178. [Google Scholar] [CrossRef]
- Reyes, N.; Onadeko, O.O.; Luraschi-Monjagatta, M.d.C.; Knox, K.S.; Rennels, M.A.; Walsh, T.K.; Ampel, N.M. Positron emission tomography in the evaluation of pulmonary nodules among patients living in a coccidioidal endemic region. Lung 2014, 192, 589–593. [Google Scholar] [CrossRef]
- Sarosi, G.A.; Parker, J.D.; Doto, I.L.; Tosh, F.E. Chronic Pulmonary Coccidioidomycosis. N. Engl. J. Med. 1970, 283, 325–329. [Google Scholar] [CrossRef]
- Tsang, C.A.; Anderson, S.M.; Imholte, S.B.; Erhart, L.M.; Chen, S.; Park, B.J.; Christ, C.; Komatsu, K.K.; Chiller, T.; Sunenshine, R.H. Enhanced surveillance of coccidioidomycosis, Arizona, USA, 2007–2008. Emerg. Infect. Dis. 2010, 16, 1738–1744. [Google Scholar]
- Bays, D.J.; Thompson, G.R.; Reef, S.; Snyder, L.; Freifeld, A.J.; Huppert, M.; Salkin, D.; Wilson, M.D.; Galgiani, J.N. Natural History of Disseminated Coccidioidomycosis: Examination of the Veterans Affairs-Armed Forces Database. Clin. Infect. Dis. 2021, 73, e3814–e3819. [Google Scholar] [CrossRef]
- Vincent, T.; Galgiani, J.N.; Huppert, M.; Salkin, D. The Natural History of Coccidioidal Meningitis: VA-Armed Forces Cooperative Studies, 1955–1958. Clin. Infect. Dis. 1993, 16, 247–254. [Google Scholar] [CrossRef]
- Einstein, H.E.; Holeman, C.W., Jr.; Sandidge, L.L.; Holden, D.H. Coccidioidal meningitis. The use of amphotericin B in treatment. Calif. Med. 1961, 94, 339–343. [Google Scholar]
- Odio, C.D.; Marciano, B.E.; Galgiani, J.N.; Holland, S.M. Risk Factors for Disseminated Coccidioidomycosis, United States. Emerg. Infect. Dis. 2017, 23, 4. [Google Scholar] [CrossRef] [PubMed]
- Wack, E.E.; Ampel, N.M.; Galgiani, J.N.; Bronnimann, D.A. Coccidioidomycosis during pregnancy. An analysis of ten cases among 47,120 pregnancies. Chest 1988, 94, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Galgiani, J.N.; Catanzaro, A.; Cloud, G.A.; Higgs, J.; Friedman, B.A.; Larsen, R.A.; Graybill, J.R. Fluconazole therapy for coccidioidal meningitis. The NIAID-Mycoses Study Group. Ann. Intern. Med. 1993, 119, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Graybill, J.R.; Stevens, D.A.; Galgiani, J.N.; Dismukes, W.E.; Cloud, G.A. Itraconazole treatment of coccidioidomycosis. NAIAD Mycoses Study Group. Am. J. Med. 1990, 89, 282–290. [Google Scholar] [CrossRef]
- Catanzaro, A.; Cloud, G.A.; Stevens, D.A.; Levine, B.E.; Williams, P.L.; Johnson, R.H.; Rendon, A.; Mirels, L.F.; Lutz, J.E.; Holloway, M.; et al. Safety, tolerance, and efficacy of posaconazole therapy in patients with nonmeningeal disseminated or chronic pulmonary coccidioidomycosis. Clin. Infect. Dis. 2007, 45, 562–568. [Google Scholar] [CrossRef]
- Pappagianis, D. Epidemiology of coccidioidomycosis. Curr. Top. Med. Mycol. 1988, 2, 199–238. [Google Scholar]
- Pappagianis, D.; Lindsay, S.; Beall, S.; Williams, P. Ethnic background and the clinical course of coccidioidomycosis [letter]. Am. Rev. Respir. Dis. 1979, 120, 959–961. [Google Scholar]
- Huppert, M. Racism in coccidioidomycosis? Am. Rev. Respir. Dis. 1978, 118, 797–798. [Google Scholar] [CrossRef]
- Kittles, R. Nature, Origin, and Variation of Human Pigmentation. J. Black Stud. 2016, 26, 36–61. [Google Scholar] [CrossRef]
- Torres, J.B.; Kittles, R.A. The relationship between "race" and genetics in biomedical research. Curr. Hypertens. Rep. 2007, 9, 196–201. [Google Scholar] [CrossRef]
- Deresinski, S.C.; Pappagianis, D.; Stevens, D.A. Association of ABO blood group and outcome of coccidioidal infection. Sabouraudia 1979, 17, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.M.; Galgiani, J.N.; Potter, D.; Ogden, D.A. Coccidioidomycosis in renal replacement therapy. Arch. Intern. Med. 1982, 142, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Mourant, A.E.; Kopec, A.C.; Kazmiera, D.S. The ABO Blood Groups; Blackwell Scientific Publications: Oxford, UK, 1959. [Google Scholar]
- Flynn, N.M.; Hoeprich, P.D.; Kawachi, M.M.; Lee, K.K.; Lawrence, R.M.; Goldstein, E.; Jordan, G.W.; Kundargi, R.S.; Wong, G.A. An unusual outbreak of windborne coccidioidomycosis. N. Engl. J. Med. 1979, 301, 358–361. [Google Scholar] [CrossRef] [PubMed]
- McHardy, I.; Reagan, K.L.; Sebastian, J.F.; Barker, B.; Bays, D.J.; Dandekar, S.; Cohen, S.H.; Jennings, K.E.; Sykes, J.; Thompson, G.R. Sex Differences in the Susceptibility to Coccidioidomycosis. Open Forum Infect. Dis. 2022, 9, ofab543. [Google Scholar]
- Kirkland, T.N.; Fierer, J. Inbred Mouse Strains Differ in Resistance to Lethal Coccidioides immitis Infection. Infect. Immun. 1983, 40, 912–916. [Google Scholar] [CrossRef]
- Shubitz, L.F.; Dial, S.M.; Perrill, R.; Casement, R.; Galgiani, J.N. Vaccine-induced cellular immune responses differ from innate responses in susceptible and resistant strains of mice infected with Coccidioides posadasii. Infect. Immun. 2008, 76, 5553–5564. [Google Scholar] [CrossRef] [PubMed]
- Fierer, J.; Walls, L.; Wright, F.; Kirkland, T.N. Genes influencing resistance to Coccidioides immitis and the interleukin-10 response map to chromosomes 4 and 6 in mice. Infect. Immun. 1999, 67, 2916–2919. [Google Scholar] [CrossRef]
- Viriyakosol, S.; Fierer, J.; Brown, G.D.; Kirkland, T.N. Innate immunity to the pathogenic fungus Coccidioides posadasii is dependent on Toll-like receptor 2 and Dectin-1. Infect. Immun. 2005, 73, 1553–1560. [Google Scholar]
- del Pilar Jimenez, A.M.; Viriyakosol, S.; Walls, L.; Datta, S.K.; Kirkland, T.; Heinsbroek, S.E.; Brown, G.; Fierer, J. Susceptibility to Coccidioides species in C57BL/6 mice is associated with expression of a truncated splice variant of Dectin-1 (Clec7a). Genes Immun. 2008, 9, 338–348. [Google Scholar] [CrossRef]
- Friedman, L.; Smith, C.E.; Roessler, W.G.; Berman, R.J. The virulence and infectivity of twenty-seven strains of Coccidioides immitis. Am. J. Hyg. 1956, 64, 198–210. [Google Scholar]
- Muhammed, M.; Feldmesser, M.; Shubitz, L.F.; Lionakis, M.S.; Sil, A.; Wang, Y.; Glavis-Bloom, J.; Lewis, R.E.; Galgiani, J.N.; Casadevall, A. Mouse models for the study of fungal pneumonia: A collection of detailed experimental protocols for the study of Coccidioides, Cryptococcus, Fusarium, Histoplasma and combined infection due to Aspergillus-Rhizopus. Virulence 2012, 3, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Shubitz, L.F.; Powell, D.A.; Butkiewicz, C.D.; Lewis, M.L.; Trinh, H.T.; Frelinger, J.A.; Orbach, M.J.; Galgiani, J.N. A Chronic Murine Disease Model of Coccidioidomycosis Using Coccidioides posadasii, Strain 1038. J. Infect. Dis. 2021, 223, 166–173. [Google Scholar] [CrossRef]
- Hsu, A.P.; Korzeniowska, A.; Aguilar, C.C.; Gu, J.; Karlins, E.; Oler, A.J.; Chen, G.; Reynoso, G.V.; Davis, J.; Chaput, A.; et al. Immunogenetics associated with severe coccidioidomycosis. JCI Insight. 2022, 7, 15. [Google Scholar] [CrossRef]
- Galgiani, J.N.; Blair, J.E.; Ampel, N.M.; Thompson, G.R. Treatment for Early, Uncomplicated Coccidioidomycosis: What Is Success? Clin. Infect. Dis. 2020, 70, 2008–2012. [Google Scholar] [CrossRef]
- Lee, L.A.; Sondermeyer Cooksey, G.L.; Kim, J.J.; Kahal, A.; Gilliss, D.; Naeem, F.; McCarty, J.M.; Vugia, D.J. Pediatric Coccidioidomycosis: Case Series From a California Pediatric Infectious Diseases Clinic. Pediatr. Infect. Dis. J. 2019, 38, 115–121. [Google Scholar] [CrossRef]
- Santelli, A.C.; Blair, J.E.; Roust, L.R. Coccidioidomycosis in patients with diabetes mellitus. Am. J. Med. 2006, 119, 964–969. [Google Scholar] [CrossRef]
- Sandler, M. Is the lung a ‘target organ’ in diabetes mellitus. Arch. Intern. Med. 1990, 150, 1385–1388. [Google Scholar] [CrossRef]
- Powell, D.A.; Hsu, A.P.; Shubitz, L.F.; Butkiewicz, C.D.; Moale, H.; Trinh, H.T.; Doetschman, T.; Georgieva, T.G.; Reinartz, D.M.; Wilson, J.E.; et al. Mouse Model of a Human STAT4 Point Mutation That Predisposes to Disseminated Coccidiomycosis. Immunohorizons 2022, 6, 130–143. [Google Scholar] [CrossRef]
- Smith, C.E.; Beard, R.R.; Saito, M.T. Pathogenesis of coccidioidomycosis with special reference to pulmonary cavitation. Ann. Intern. Med. 1948, 29, 623–655. [Google Scholar]
- Adam, R.D.; Elliott, S.P.; Taljanovic, M.S. The spectrum and presentation of disseminated coccidioidomycosis. Am. J. Med. 2009, 122, 770–777. [Google Scholar] [CrossRef]
- Smith, C.E.; Pappagianis, D.; Levine, H.B.; Saito, M.T. Human coccidioidomycosis. Bacteriol. Rev. 1961, 25, 310–320. [Google Scholar] [CrossRef]
- Forseth, J.; Rohwedder, J.J.; Levine, B.E.; Saubolle, M.A. Experience with needle biopsy for coccidioidal lung nodules. Arch. Intern. Med. 1986, 146, 319–320. [Google Scholar] [CrossRef]
- Chitkara, Y.K. Evaluation of cultures of percutaneous core needle biopsy specimens in the diagnosis of pulmonary nodules. Am. J. Clin. Pathol. 1997, 107, 224–228. [Google Scholar] [CrossRef]
- Deresinski, S.C.; Stevens, D.A. Coccidioidomycosis in compromised hosts. Experience at Stanford University Hospital. Medicine 1974, 54, 377–395. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.L.; Fleming, P.L.; Ciesielski, C.A.; Hu, D.J.; Kaplan, J.E.; Ward, J.W. Coccidioidomycosis among persons with AIDS in the United States. J. Infect. Dis. 1995, 171, 961–966. [Google Scholar] [CrossRef]
- Hernandez, J.L.; Echevarria, S.; Garcia-Valtuille, A.; Mazorra, F.; Salesa, R. Atypical coccidioidomycosis in an AIDS patient successfully treated with fluconazole. Eur. J. Clin. Microbiol. Infect. Dis. 1997, 16, 592–594. [Google Scholar] [CrossRef]
- Bergstrom, L.; Yocum, D.E.; Ampel, N.M.; Villanueva, I.; Lisse, J.; Gluck, O.; Tesser, J.; Posever, J.; Miller, M.; Araujo, J.; et al. Increased risk of coccidioidomycosis in patients treated with tumor necrosis factor alpha antagonists. Arthritis Rheum. 2004, 50, 1959–1966. [Google Scholar] [CrossRef]
- Taroumian, S.; Knowles, S.L.; Lisse, J.R.; Yanes, J.; Ampel, N.M.; Vaz, A.; Galgiani, J.N.; Hoover, S.E. Management of coccidioidomycosis in patients receiving biologic response modifiers or disease-modifying antirheumatic drugs. Arthritis Care Res. 2012, 64, 1903–1909. [Google Scholar] [CrossRef]
- Donovan, F.M.; Ramadan, F.A.; Lim, J.R.; Buchfuhrer, J.E.; Khan, R.N.; DeQuillfeldt, N.P.; Davis, N.M.; Kaveti, A.; De Shadarevian, M.; Bedrick, E.J.; et al. Contribution of Biologic Response Modifiers to the Risk of Coccidioidomycosis Severity. Open Forum. Infect. Dis. 2022, 9, ofac032. [Google Scholar]
- Wallis, R.S. Mycobacterial disease attributable to tumor necrosis factor-alpha blockers. Clin. Infect. Dis. 2008, 47, 1603–1605. [Google Scholar] [CrossRef]
- Powell, D.A.; Shubitz, L.F.; Butkiewicz, C.D.; Trinh, H.T.; Donovan, F.M.; Frelinger, J.A.; Galgiani, J.N. TNF-alpha Blockade Inhibits Both Initial and Continued Control of Pulmonary Coccidioides. Front. Cell Infect. Microbiol. 2021, 11, 796114. [Google Scholar] [CrossRef]
- Fréling, E.; Baumann, C.; Cuny, J.-F.; Bigard, M.-A.; Schmutz, J.-L.; Barbaud, A.; Peyrin-Biroulet, L. Cumulative Incidence of, Risk Factors for, and Outcome of Dermatological Complications of Anti-TNF Therapy in Inflammatory Bowel Disease: A 14-Year Experience. Off. J. Am. Coll. Gastroenterol. 2015, 110, 1186–1196. [Google Scholar] [CrossRef]
- van Dartel, S.A.; Fransen, J.; Kievit, W.; Flendrie, M.; den Broeder, A.A.; Visser, H.; Hartkamp, A.; van de Laar, M.A.; van Riel, P.L. Difference in the risk of serious infections in patients with rheumatoid arthritis treated with adalimumab, infliximab and etanercept: Results from the Dutch Rheumatoid Arthritis Monitoring (DREAM) registry. Ann. Rheum. Dis. 2013, 72, 895–900. [Google Scholar] [CrossRef]
- Crum, N.F.; Lederman, E.R.; Wallace, M.R. Infections Associated with Tumor Necrosis Factor-α Antagonists. Medicine 2005, 84, 291–302. [Google Scholar] [CrossRef]
- Catanzaro, A.; Galgiani, J.N.; Levine, B.E.; Sharkey-Mathis, P.K.; Fierer, J.; Stevens, D.A.; Chapman, S.W.; Cloud, G.; NIAID Mycoses Study Group. Fluconazole in the treatment of chronic pulmonary and nonmeningeal disseminated coccidioidomycosis. Am. J. Med. 1995, 98, 249–256. [Google Scholar] [CrossRef]
- Dewsnup, D.H.; Galgiani, J.N.; Graybill, J.R.; Diaz, M.; Rendon, A.; Cloud, G.A.; Stevens, D.A. Is it ever safe to stop azole therapy for Coccidioides immitis meningitis? Ann. Intern. Med. 1996, 124, 305–310. [Google Scholar]
- Petersen, E.A.; Friedman, B.A.; Crowder, E.D.; Rifkind, D. Coccidoidouria: Clinical significance. Ann. Intern. Med. 1976, 85, 34–38. [Google Scholar] [CrossRef]
- DeFelice, R.; Wieden, M.A.; Galgiani, J.N. The incidence and implications of coccidioidouria. Am. Rev. Respir. Dis. 1982, 125, 49–52. [Google Scholar]
- Yurkanin, J.P.; Ahmann, F.; Dalkin, B.L. Coccidioidomycosis of the prostate: A determination of incidence, report of 4 cases, and treatment recommendations. J. Infect. 2006, 52, e19–e25. [Google Scholar] [CrossRef]
- Rodenbiker, H.T.; Ganley, J.P. Ocular coccidioidomycosis. Surv. Ophthalmol. 1980, 24, 263–290. [Google Scholar] [CrossRef]
- Rodenbiker, H.T.; Ganley, J.P.; Galgiani, J.N.; Axline, S.G. Prevalence of chorioretinal scars associated with coccidioidomycosis. Arch. Ophthalmol. 1981, 99, 71–75. [Google Scholar] [CrossRef]
- Oldfield, E.C., III; Bone, W.D.; Martin, C.R.; Gray, G.C.; Olson, P.; Schillaci, R.F. Prediction of relapse after treatment of coccidioidomycosis. Clin. Infect. Dis. 1997, 25, 1205–1210. [Google Scholar] [CrossRef]
- Freedman, M.; Jackson, B.R.; McCotter, O.; Benedict, K. Coccidioidomycosis Outbreaks, United States and Worldwide, 1940–2015. Emerg. Infect. Dis. 2018, 24, 417–423. [Google Scholar]
- Shin, H.; Park, S.; Lee, G.W.; Koh, E.H.; Kim, H.Y. Parvovirus B19 infection presenting with neutropenia and thrombocytopenia: Three case reports. Medicine 2019, 98, e16993. [Google Scholar] [CrossRef]
- Estrada, L.; August, J.; Ojo, T.; Campion, J. Prevalence of coccidioidomycosis in cystic fibrosis patients residing in Southern Arizona. Med. Mycol. 2021, 59, 309–312. [Google Scholar] [CrossRef]
- Gomez-Duarte, O.G.; Ogra, P.L. Editorial: Impact of microbiome on gut mucosal immunity in health and disease. Front. Immunol. 2022, 13, 1005156. [Google Scholar] [CrossRef]
- Bagnasco, D.; Caminati, M. Editorial: Innate immunity and severe asthma: From microbiome to target therapy. Front. Immunol. 2022, 13, 1114275. [Google Scholar] [CrossRef]
- Kharrazian, D. Exposure to Environmental Toxins and Autoimmune Conditions. Integr. Med. 2021, 20, 20–24. [Google Scholar]
- Magan, D.; Yadav, R.K. Psychoneuroimmunology of Meditation. Ann. Neurosci. 2022, 29, 170–176. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Study | Subjects | ||
|---|---|---|---|
| All | Immunocompromised | Male | |
| Fluconazole for meningitis [36] | 50 | 12 (24%) | 41 (82%) |
| Itraconazole for non-meningeal dissemination [37] | 26 | 1 (3.8%) | 21 (81%) |
| Posaconazole for non-meningeal dissemination [38] | 12 | 2 (18%) | 8 (67%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galgiani, J.N.; Hsu, A.P.; Powell, D.A.; Vyas, J.M.; Holland, S.M. Genetic and Other Determinants for the Severity of Coccidioidomycosis: A Clinician’s Perspective. J. Fungi 2023, 9, 554. https://doi.org/10.3390/jof9050554
Galgiani JN, Hsu AP, Powell DA, Vyas JM, Holland SM. Genetic and Other Determinants for the Severity of Coccidioidomycosis: A Clinician’s Perspective. Journal of Fungi. 2023; 9(5):554. https://doi.org/10.3390/jof9050554
Chicago/Turabian StyleGalgiani, John N., Amy P. Hsu, Daniel A. Powell, Jatin M. Vyas, and Steven M. Holland. 2023. "Genetic and Other Determinants for the Severity of Coccidioidomycosis: A Clinician’s Perspective" Journal of Fungi 9, no. 5: 554. https://doi.org/10.3390/jof9050554
APA StyleGalgiani, J. N., Hsu, A. P., Powell, D. A., Vyas, J. M., & Holland, S. M. (2023). Genetic and Other Determinants for the Severity of Coccidioidomycosis: A Clinician’s Perspective. Journal of Fungi, 9(5), 554. https://doi.org/10.3390/jof9050554