
In Vivo Microevolutionary Analysis of a Fatal Case of Rhinofacial and Disseminated Mycosis Due to Azole-Drug-Resistant Candida Species

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. All Candida Strains Culture and Antifungal Susceptibility Assays
2.2. Genome Sequencing and Assembly
2.3. Genomic Prediction and Annotation
2.4. Phylogenetic Analysis
2.5. Variant Calling
2.6. Verification of SNPs and Indels
2.7. Quantification and Statistical Analysis
2.8. Ethical Declaration
3. Results
3.1. Patient Course and Candida Strains
3.2. Genome Features and Species Identification
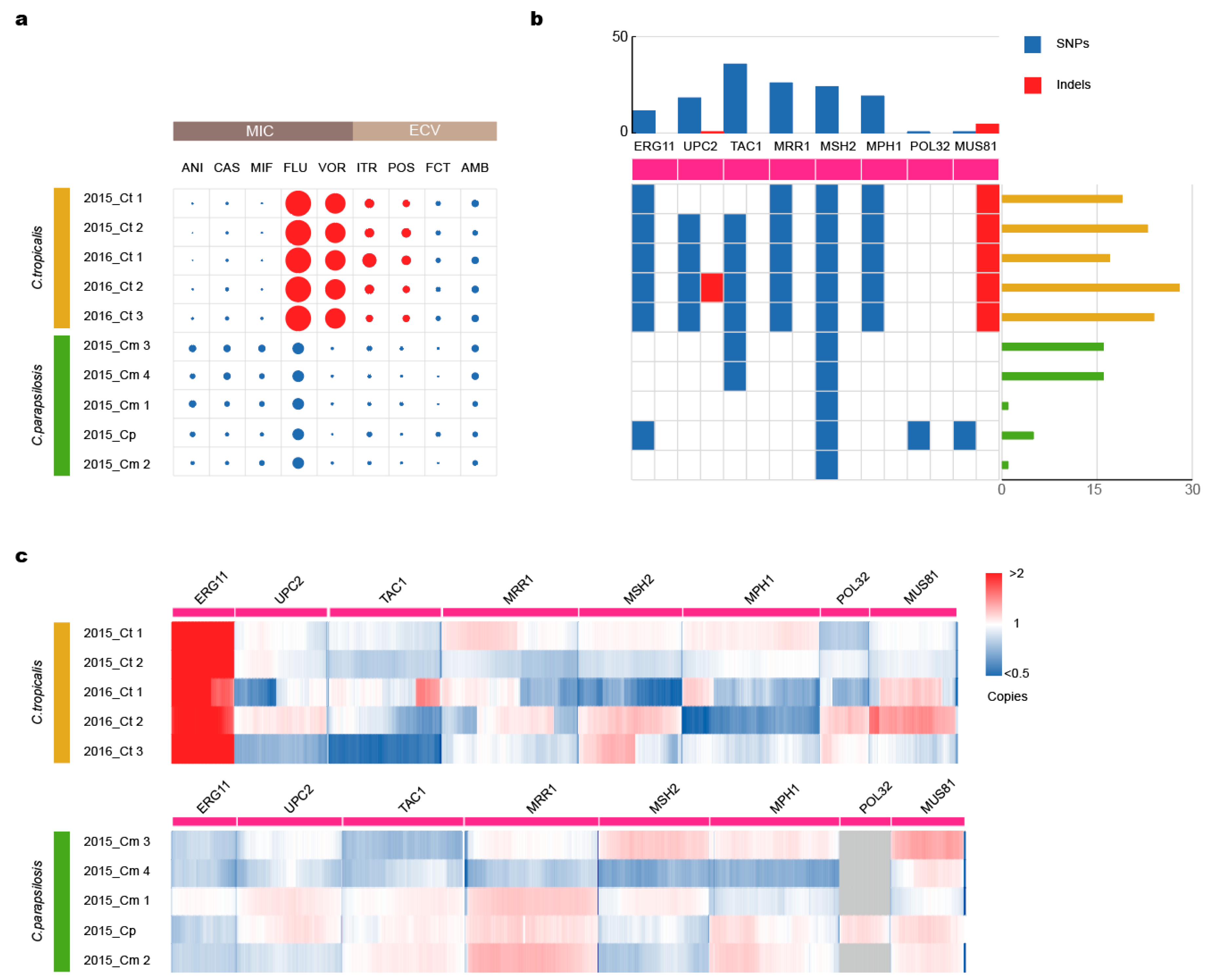
3.3. Mutations in Drug-Resistance Genes Lead to Azole Resistance
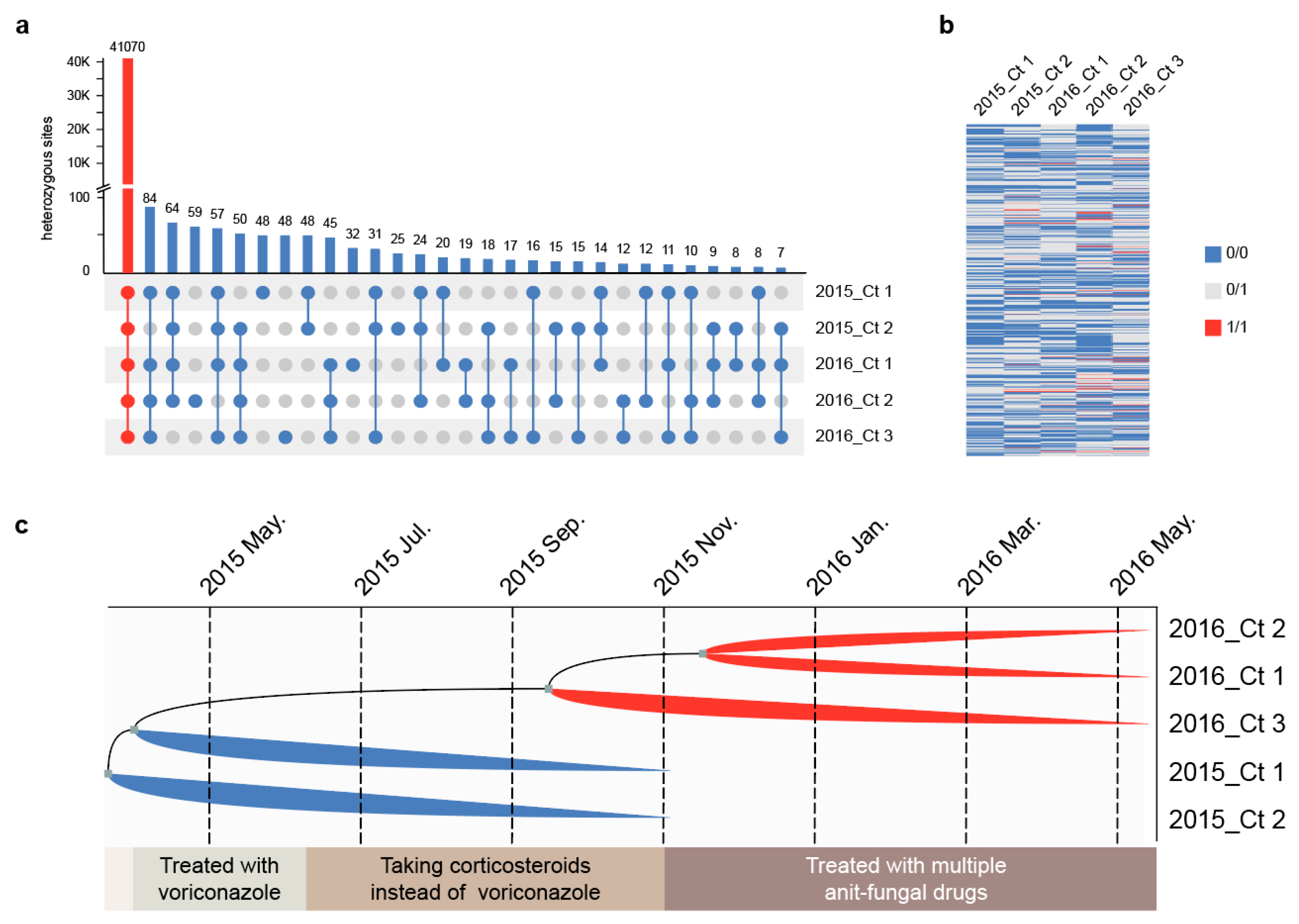
3.4. Strain Differentiation Is Associated with Antifungal Treatment
3.5. Multi-Drug Resistant Strains Originated from the Same Clone and Underwent Parallel Evolution In Vivo
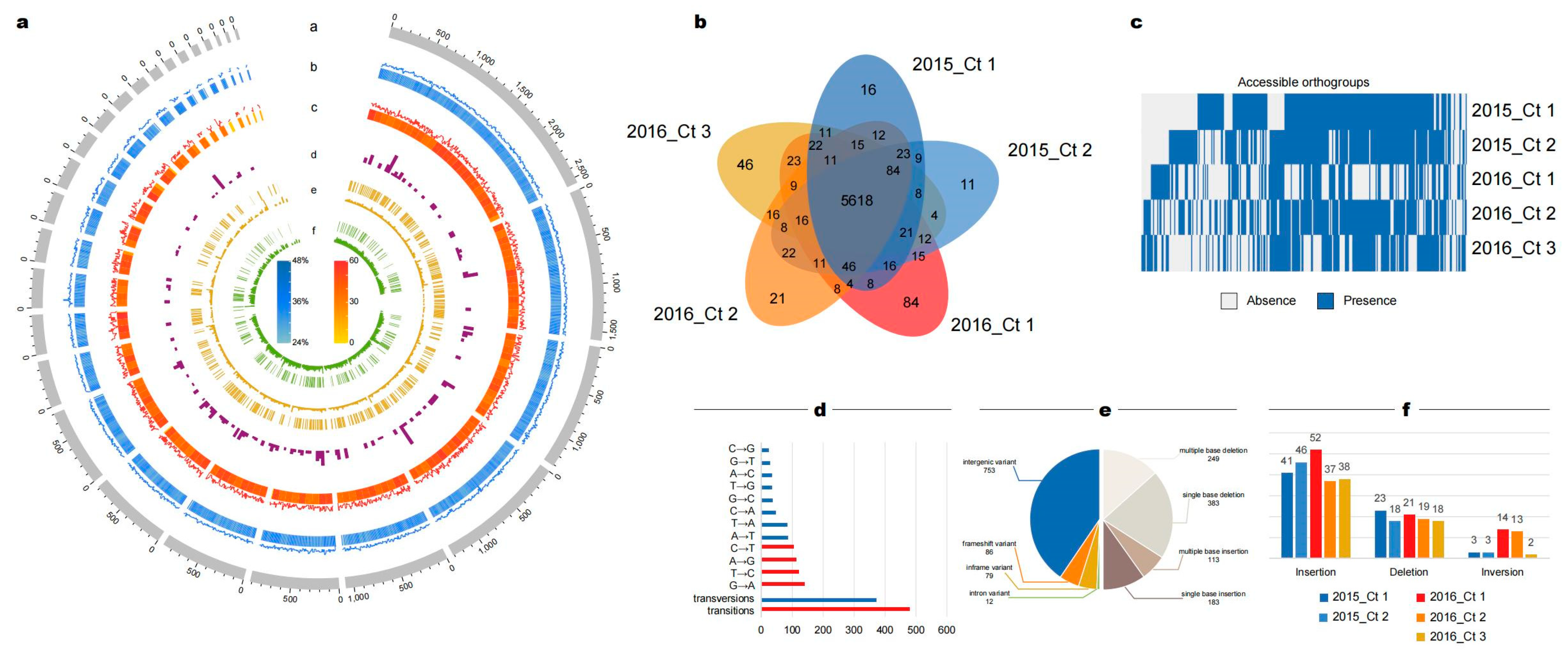
3.6. Abnormal Number of Insertions and Deletions May Be Caused by DNA Mismatch Repair Genes
3.7. Strains Isolated after Multiple Antifungal Administrations Exhibited Decreased Virulence
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassetti, M.; Peghin, M.; Timsit, J.F. The current treatment landscape: Candidiasis. J. Antimicrob. Chemother. 2016, 71 (Suppl. 2), ii13–ii22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Ding, Y.; Jiang, Y.; Mo, S.; Xu, S.; Qin, P. Persistent candidemia in very low birth weight neonates: Risk factors and clinical significance. BMC Infect. Dis. 2018, 18, 558. [Google Scholar] [CrossRef]
- Xiao, M.; Chen, S.C.; Kong, F.; Xu, X.L.; Yan, L.; Kong, H.S.; Fan, X.; Hou, X.; Cheng, J.W.; Zhou, M.L.; et al. Distribution and antifungal susceptibility of Candida species causing candidemia in China: An update from the CHIF-NET study. J. Infect. Dis. 2020, 221 (Suppl. 2), S139–S147. [Google Scholar] [CrossRef]
- Da Matta, D.A.; Souza, A.C.R.; Colombo, A.L. Revisiting Species Distribution and Antifungal Susceptibility of Candida Bloodstream Isolates from Latin American Medical Centers. J. Fungi 2017, 3, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, L.N.; de Mello, T.P.; de Souza Ramos, L.; Branquinha, M.H.; Dos Santos, A.L.S. New and promising chemotherapeutics for emerging infections involving drug-resistant non-albicans Candida species. Curr. Top. Med. Chem. 2019, 19, 2527–2553. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, J.A.; Hu, C.; Aitken, S.L.; Beyda, N. Antifungal resistance: A concerning trend for the present and future. Curr. Infect. Dis. Rep. 2019, 21, 47. [Google Scholar] [CrossRef]
- Ksiezopolska, E.; Gabaldón, T. Evolutionary emergence of drug resistance in candida opportunistic pathogens. Genes 2018, 9, 461. [Google Scholar] [CrossRef] [Green Version]
- Arastehfar, A.; Daneshnia, F.; Hafez, A.; Khodavaisy, S.; Najafzadeh, M.-J.; Charsizadeh, A.; Zarrinfar, H.; Salehi, M.; Shahrabadi, Z.Z.; Sasani, E.; et al. Antifungal susceptibility, genotyping, resistance mechanism, and clinical profile of Candida tropicalis blood isolates. Mycology 2020, 58, 766–773. [Google Scholar] [CrossRef]
- Fisher, M.C.; Alastruey-Izquierdo, A.; Berman, J.; Bicanic, T.; Bignell, E.M.; Bowyer, P.; Bromley, M.; Brüggemann, R.; Garber, G.; Cornely, O.A.; et al. Tackling the emerging threat of antifungal resistance to human health. Nat. Rev. Microbiol. 2022, 20, 557–571. [Google Scholar] [CrossRef]
- Arendrup, M.C.; Patterson, T.F. Multidrug-resistant Candida: Epidemiology, molecular mechanisms, and treatment. J. Infect. Dis. 2017, 216 (Suppl. 3), S445–S451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos-Garzon, A.; Wintaco-Martínez, L.M.; Velez, N.; Hernandez-Padilla, C.; De la Hoz, A.; Valderrama-Beltrán, S.L.; Alvarez-Moreno, C.A.; Pape, P.L.; Ramírez, J.D.; Parra-Giraldo, C.M. Persistence of Clonal Azole-Resistant Isolates of Candida albicans from a Patient with Chronic Mucocutaneous Candidiasis in Colombia. J. Glob. Infect. Dis. 2020, 12, 16–20. [Google Scholar] [CrossRef]
- Khosravi, A.R.; Mansouri, P.; Saffarian, Z.; Vahedi, G.; Nikaein, D. Chronic mucocutaneous candidiasis, a case study and literature review. J. Mycol. Med. 2018, 28, 206–210. [Google Scholar] [CrossRef]
- Okada, S.; Puel, A.; Casanova, J.L.; Kobayashi, M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin. Transl. Immunol. 2016, 5, e114. [Google Scholar] [CrossRef]
- Vaughan, C.; Bartolo, A.; Vallabh, N.; Leong, S.C. A meta-analysis of survival factors in rhino-orbital-cerebral mucormycosis-has anything changed in the past 20 years? Clin. Otolaryngol. 2018, 43, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.B.; Li, D.M.; Houbraken, J.; Sun, T.T.; de Hoog, G.S. Fatal Rhinofacial Mycosis Due to Aspergillus nomiae: Case Report and Review of Published Literature. Front. Microbiol. 2020, 11, 595375. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, C.A. Harnessing Whole Genome Sequencing in Medical Mycology. Curr. Fungal Infect. Rep. 2017, 11, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Schikora-Tamarit, M.À.; Gabaldón, T. Using genomics to understand the mechanisms of virulence and drug resistance in fungal pathogens. Biochem. Soc. Trans. 2022, 50, 1259–1268. [Google Scholar] [CrossRef]
- Tagini, F.; Greub, G. Bacterial genome sequencing in clinical microbiology: A pathogen-oriented review. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2007–2020. [Google Scholar] [CrossRef] [PubMed]
- Salem-Bango, Z.; Price, T.K.; Chan, J.L.; Chandrasekaran, S.; Garner, O.B.; Yang, S. Fungal Whole-Genome Sequencing for Species Identification: From Test Development to Clinical Utilization. J. Fungi 2023, 9, 183. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, 4th ed.; Approved standard M27; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017. [Google Scholar]
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi; Approved Standard; CLSI Document m38-A2; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017. [Google Scholar]
- Clinical and Laboratory Standards Institute. Epidemiological Cutoff Values for Antifungal Susceptibility Testing, 2nd ed.; CLSI supplement M59; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]
- Liu, J.; Guo, L.; Li, Z.; Zhou, Z.; Li, Z.; Li, Q.; Bo, X.; Wang, S.; Wang, J.; Ma, S.; et al. Genomic analyses reveal evolutionary and geologic context for the plateau fungus Ophiocordyceps sinensis. Chin. Med. 2020, 15, 107. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, M.; Li, Z.; Al-Hatmi, A.M.; de Hoog, S.; Pan, W.; Ye, Q.; Bo, X.; Li, Z.; Wang, S.; et al. Genome sequencing and comparative genomics analysis revealed pathogenic potential in Penicillium capsulatum as a novel fungal pathogen belonging to Eurotiales. Front. Microbiol. 2016, 7, 1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Keller, O.; Kollmar, M.; Stanke, M.; Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 2011, 27, 757–763. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Hong, N.; Hu, S.; Wang, P.; Guan, H.; Xiao, M.; Zhu, X.; Al-Hatmi, A.M.S.; Zhou, Z.; Gao, L.; et al. Molecular identification of Cryptococcus gattii from cerebrospinal fluid using single-cell sequencing: A case study. J. Infect. 2020, 81, 634–638. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis, version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.H.; Guo, H.; Wang, X.; Kim, C.; Paterson, A.H. SNPhylo: A pipeline to construct a phylogenetic tree from huge SNP data. BMC Genom. 2014, 15, 162. [Google Scholar] [CrossRef] [Green Version]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P. Variant annotation and functional prediction: SnpEff. Methods Mol. Biol. 2022, 2493, 289–314. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ye, Q.; Li, K.; Li, Z.; Bo, X.; Li, Z.; Xu, Y.; Wang, S.; Wang, P.; Chen, H.; et al. Genomics and comparative genomic analyses provide insight into the taxonomy and pathogenic potential of novel Emmonsia pathogens. Front. Cell. Infect. Microbiol. 2017, 7, 105. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Jiang, C.; Dong, D.; Yu, B.; Cai, G.; Wang, X.; Ji, Y.; Peng, Y. Mechanisms of azole resistance in 52 clinical isolates of Candida tropicalis in China. J. Antimicrob. Chemother. 2013, 68, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Zheng, Q.; Bing, J.; Bennett, R.J.; Huang, G. A coupled process of same- and opposite-sex mating generates polyploidy and genetic diversity in Candida tropicalis. PLoS Genet. 2018, 14, e1007377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 1969, 61, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Gregory, I. Lang and others, Mutation Rates, Spectra, and Genome-Wide Distribution of Spontaneous Mutations in Mismatch Repair Deficient Yeast. G3 Genes Genomes Genet. 2013, 3, 1453–1465. [Google Scholar] [CrossRef] [Green Version]
- Pryszcz, L.P.; Németh, T.; Saus, E.; Ksiezopolska, E.; Hegedűsová, E.; Nosek, J.; Wolfe, K.H.; Gacser, A.; Gabaldón, T. The Genomic Aftermath of Hybridization in the Opportunistic Pathogen Candida metapsilosis. PLoS Genet. 2015, 11, e1005626. [Google Scholar] [CrossRef]
- Kamai, Y.; Maebashi, K.; Kudoh, M.; Makimura, K.; Naka, W.; Uchida, K.; Yamaguchi, H. Characterization of mechanisms of fluconazole resistance in a Candida albicans isolate from a Japanese patient with chronic mucocutaneous candidiasis. Microbiol. Immunol. 2004, 48, 937–943. [Google Scholar] [CrossRef]
- Vandeputte, P.; Larcher, G.; Bergès, T.; Renier, G.; Chabasse, D.; Bouchara, J.P. Mechanisms of azole resistance in a clinical isolate of Candida tropicalis. Antimicrob. Agents Chemother. 2005, 49, 4608–4615. [Google Scholar] [CrossRef] [Green Version]
- Heilmann, C.J.; Schneider, S.; Barker, K.S.; Rogers, P.D.; Morschhäuser, J. An A643T mutation in the transcription factor Upc2p causes constitutive ERG11 upregulation and increased fluconazole resistance in Candida albicans. Antimicrob. Agents Chemother. 2010, 54, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Hoot, S.J.; Smith, A.R.; Brown, R.P.; White, T.C. An A643V amino acid substitution in Upc2p contributes to azole resistance in well-characterized clinical isolates of Candida albicans. Antimicrob. Agents Chemother. 2011, 55, 940–942. [Google Scholar] [CrossRef] [Green Version]
- Flowers, S.A.; Barker, K.S.; Berkow, E.L.; Toner, G.; Chadwick, S.G.; Gygax, S.E.; Morschhäuser, J.; Rogers, P.D. Gain-of-function mutations in UPC2 are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans. Eukaryot. Cell 2012, 11, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Tong, J.; Lee, C.W.; Ha, S.; Eom, S.H.; Im, Y.J. Structural mechanism of ergosterol regulation by fungal sterol transcription factor Upc2. Nat. Commun. 2015, 6, 6129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morschhäuser, J.; Barker, K.S.; Liu, T.T.; BlaB-Warmuth, J.; Homayouni, R.; Rogers, P.D. The transcription factor Mrr1p controls expression of the MDR1 efflux pump and mediates multidrug resistance in Candida albicans. PLoS Pathog. 2007, 3, e164. [Google Scholar] [CrossRef] [Green Version]
- Dunkel, N.; Blass, J.; Rogers, P.D.; Morschhäuser, J. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol. Microbiol. 2008, 69, 827–840. [Google Scholar] [CrossRef] [Green Version]
- Hampe, I.A.I.; Friedman, J.; Edgerton, M.; Morschhäuser, J. An acquired mechanism of antifungal drug resistance simultaneously enables Candida albicans to escape from intrinsic host defenses. PLoS Pathog. 2017, 13, e1006655. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Hull, C.M.; Heitman, J. Sexual reproduction between partners of the same mating type in Cryptococcus neoformans. Nature 2005, 434, 1017–1021. [Google Scholar] [CrossRef]
- Alby, K.; Schaefer, D.; Bennett, R.J. Homothallic and heterothallic mating in the opportunistic pathogen Candida albicans. Nature 2009, 460, 890–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xu, J. Population genomic analyses reveal evidence for limited recombination in the superbug Candida auris in nature. Comput. Struct. Biotechnol. J. 2022, 20, 3030–3040. [Google Scholar] [CrossRef]
- Tanne, J.H. Drug resistant fungus is spreading in US long term care facilities and hospitals, warns CDC. BMJ 2023, 380, 709. [Google Scholar] [CrossRef]
- Rocchi, S.; Sewell, T.R.; Valot, B.; Godeau, C.; Laboissiere, A.; Millon, L.; Fisher, M.C. Molecular Epidemiology of Azole-Resistant Aspergillus fumigatus in France Shows Patient and Healthcare Links to Environmentally Occurring Genotypes. Front. Cell Infect. Microbiol. 2021, 11, 729476. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xu, Y.; Hong, N.; Yang, Y.; Lei, W.; Du, L.; Zhao, J.; Lei, X.; Xiong, L.; Cai, L.; et al. Epidemiology of fungal infections in China. Front. Med. 2018, 12, 58–75. [Google Scholar] [CrossRef]
- Marton, T.; Chauvel, M.; Feri, A.; Maufrais, C.; D’enfert, C.; Legrand, M. Factors that influence bidirectional long-tract homozygosis due to double-strand break repair in Candida albicans. Genetics 2021, 218, iyab028. [Google Scholar] [CrossRef]
- Billmyre, R.B.; Applen Clancey, S.; Li, L.X.; Doering, T.L.; Heitman, J. 5-fluorocytosine resistance is associated with hypermutation and alterations in capsule biosynthesis in Cryptococcus. Nat. Commun. 2020, 11, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenwyk, J.L. Evolutionary Divergence in DNA Damage Responses among Fungi. MBio 2021, 16, e03348-20. [Google Scholar] [CrossRef] [PubMed]
- Whaley, S.G.; Berkow, E.L.; Rybak, J.M.; Nishimoto, A.T.; Barker, K.S.; Rogers, P.D. Azole antifungal resistance in Candida albicans and emerging non-albicans Candida species. Front. Microbiol. 2016, 7, 2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Liao, M.; Zhu, C.; Hu, Y.; Tong, T.; Peng, X.; Li, M.; Feng, M.; Cheng, L.; Ren, B.; et al. ERG3 and ERG11 genes are critical for the pathogenesis of Candida albicans during the oral mucosal infection. Int. J. Oral Sci. 2018, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Hwang, C.S.; Rhie, G.E.; Oh, J.H.; Huh, W.K.; Yim, H.S.; Kang, S.O. Copper- and zinc-containing superoxide dismutase (Cu/ZnSOD) is required for the protection of Candida albicans against oxidative stresses and the expression of its full virulence. Microbiology 2002, 148, 3705–3713. [Google Scholar] [CrossRef] [Green Version]
- Umeyama, T.; Kaneko, A.; Watanabe, H.; Hirai, A.; Uehara, Y.; Niimi, M.; Azuma, M. Deletion of the CaBIG1 gene reduces β-1,6-Glucan synthesis, filamentation, adhesion, and virulence in Candida albicans. Infect. Immun. 2006, 74, 2373–2381. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.J.; Chang, Y.L.; Chen, Y.L. Deletion of ADA2 Increases antifungal Drug Susceptibility and Virulence in Candida glabrata. Antimicrob. Agents Chemother. 2018, 62, e01924-17. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Palecek, S.P. EAP1, a Candida albicans Gene involved in binding human epithelial cells. Eukaryot. Cell 2003, 2, 1266–1273. [Google Scholar] [CrossRef] [Green Version]
- Bassilana, M.; Blyth, J.; Arkowitz, R.A. Cdc24, the GDP-GTP exchange factor for Cdc42, is required for invasive hyphal growth of Candida albicans. Eukaryot. Cell 2003, 2, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Duan, Y.; Qin, Y.; Sun, W.; Zhuang, Z.; Zhu, D.; Jiang, L. The N-terminal pY33XL motif of CaPsy2 is critical for the function of protein phosphatase 4 in CaRad53 deactivation, DNA damage-induced filamentation and virulence in Candida albicans. Int. J. Med. Microbiol. 2017, 307, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.G.; Lionakis, M.S.; Arendrup, M.C.; Ostrosky-Zeichner, L.; Kullberg, B.J. Invasive candidiasis. Nat. Rev. Dis. Primers 2018, 4, 18026. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Guo, X.; Zhang, X.; Chen, P.; Wang, W.; Hu, S.; Ma, T.; Zhou, X.; Li, D.; Yang, Y. In Vivo Microevolutionary Analysis of a Fatal Case of Rhinofacial and Disseminated Mycosis Due to Azole-Drug-Resistant Candida Species. J. Fungi 2023, 9, 815. https://doi.org/10.3390/jof9080815
Wang Y, Guo X, Zhang X, Chen P, Wang W, Hu S, Ma T, Zhou X, Li D, Yang Y. In Vivo Microevolutionary Analysis of a Fatal Case of Rhinofacial and Disseminated Mycosis Due to Azole-Drug-Resistant Candida Species. Journal of Fungi. 2023; 9(8):815. https://doi.org/10.3390/jof9080815
Chicago/Turabian StyleWang, Yuchen, Xi Guo, Xinran Zhang, Ping Chen, Wenhui Wang, Shan Hu, Teng Ma, Xingchen Zhou, Dongming Li, and Ying Yang. 2023. "In Vivo Microevolutionary Analysis of a Fatal Case of Rhinofacial and Disseminated Mycosis Due to Azole-Drug-Resistant Candida Species" Journal of Fungi 9, no. 8: 815. https://doi.org/10.3390/jof9080815