1. Introduction
Classically, cancer research has relied both on two-dimensional (2D) cell culture and on animal models. Animal models are a valuable tool in cancer research. However, they do not represent the behavior of naturally occurring cancers in humans and are expensive, time-consuming, and not feasible for high-throughput screening (HTS) [
1,
2,
3]. On the other hand, 2D in vitro cancer models are under highly controlled conditions and are highly reproducible, which make them very attractive to be used routinely for many research groups and the pharmaceutical industry. Nevertheless, it is widely accepted that 2D models do not represent the in vivo scenario, since cells are grown in a solid and flat support. Thus, cells are forced to polarize, which leads to excessive nutrition and oxygenation, and besides, molecular gradients found in vivo are not reproduced. Moreover, when culturing cells in 2D, the extracellular matrix (ECM)’s composition and configuration are strongly modified, and consequently, cells do not receive the proper signals that provide a normal ECM configuration [
4].
Cancer cells, as well as normal cells, require cues from a three-dimensional (3D) environment to form tissue structures in vitro. Moreover, tumor behavior can only be explained considering the contribution of its microenvironment, which includes the ECM (a network of fibrous proteins and proteoglycans), diffusible cytokines and growth factors, the surrounding blood vessels, as well as a set of non-cancer cells, such as fibroblasts. The tumor microenvironment plays an important role in the biology and function of cancer cells, since it provides the surrounding milieu that cancer cells need for survival, growth, proliferation, and metastasis [
5,
6,
7]. Since the microenvironment has a critical role in tumorigenesis, ECM analogs, also called “scaffolds”, have been introduced in 3D cell culture systems. In these systems, cells are embedded in a natural or synthetic scaffold that mimics the ECM, providing thus the chemical, mechanical, and physical cues that cells need to form physiological tissue structures in vitro.
In particular, the self-assembling peptide RAD16-I has been used in the present work as a scaffold for 3D cell culture. RAD16-I is a short peptide constituted by the sequence AcN-(RADA)
4-CONH
2, which alternates hydrophilic and hydrophobic amino acids [
8,
9]. The peptide undergoes self-assembly into a nanofiber network with an antiparallel β-sheet configuration by increasing ionic strength or adjusting pH to neutrality. The nanoscale architecture of the fiber network (around 10 nm diameter and 50–200 nm pore size, a thousand times smaller than mammalian cells) allows the cells to experiment in a truly 3D environment. Besides, biomolecules in such a nanoscale environment diffuse slowly and are likely to establish a local molecular gradient. Non-covalent interactions allow for cell growth, migration, contact with other cells, shape changes, and a proper exposition of membrane receptors [
10]. Moreover, since stiffness can be controlled by changing peptide concentration, these hydrogels can be tuned up to embed cells but not to entrap them [
9,
10]. Since the peptide scaffold does not contain signaling motifs, the environment can be considered non-instructive, from the point of view of cell receptor recognition/activation, making the use of this biomaterial more challenging, since factors that drive tumor progression need to be identified and precisely incorporated. On the other hand, the self-assembling peptide scaffold RAD16-I can be functionalized with signaling motifs, such as ECM ligands for cell receptors, to trigger different cellular responses [
11,
12].
The aim of the present work is to develop a 3D co-culture system, including not only cancer cells but also stromal cells, to study pancreatic ductal adenocarcinoma (PDAC), which is one of the most aggressive carcinomas [
13]. For that, pancreatic ductal adenocarcinoma cells (PANC-1), a well-characterized pancreatic cancer cell line [
14], will be co-cultured with human normal dermal fibroblasts (hNDF). With this system, is our intention to recreate a more realistic scenario in which cancer cells would receive from the 3D environment the mechanical and physical signals needed to undergo their biological functions while stromal cells will contribute with the biological signals (such as growth factors and cytokines) that could modulate tumorigenesis. Moreover, since the disadvantage of many anticancer drugs is their non-specificity cell-type activity, co-culture systems are a valuable tool to study the cytotoxic effect of anticancer drugs on normal cells. As proof of principle, the drugs Gemcitabine, 5-Fluorouracil (5-FU), and 4-Methyllumbilliferone (4-MU) will be tested on the developed 3D co-culture system. Gemcitabine and 5-FU block cell-cycle progression at G
1/S phase, targeting highly proliferating cells [
15,
16]. On the other hand, 4-MU is a potent inhibitor of hyaluronan synthesis, which has been demonstrated to decrease cell viability, migration, and invasion on different cancer cell types [
17,
18,
19,
20,
21]. Experimental results showed that Gemcitabine and 5-FU prevented PANC-1 cell proliferation but also had a high cytotoxic effect on fibroblasts. While 4-MU had a subtle effect on PANC-1 cells, it caused high cell death on fibroblasts. With this 3D co-culture system, we were able to evaluate the anticancer effect of these drugs on tumor cells and cytotoxicity on normal cells at the same time, a fact that is valuable given the non-specificity of many anticancer drugs.
3. Materials and Methods
3.1. Materials
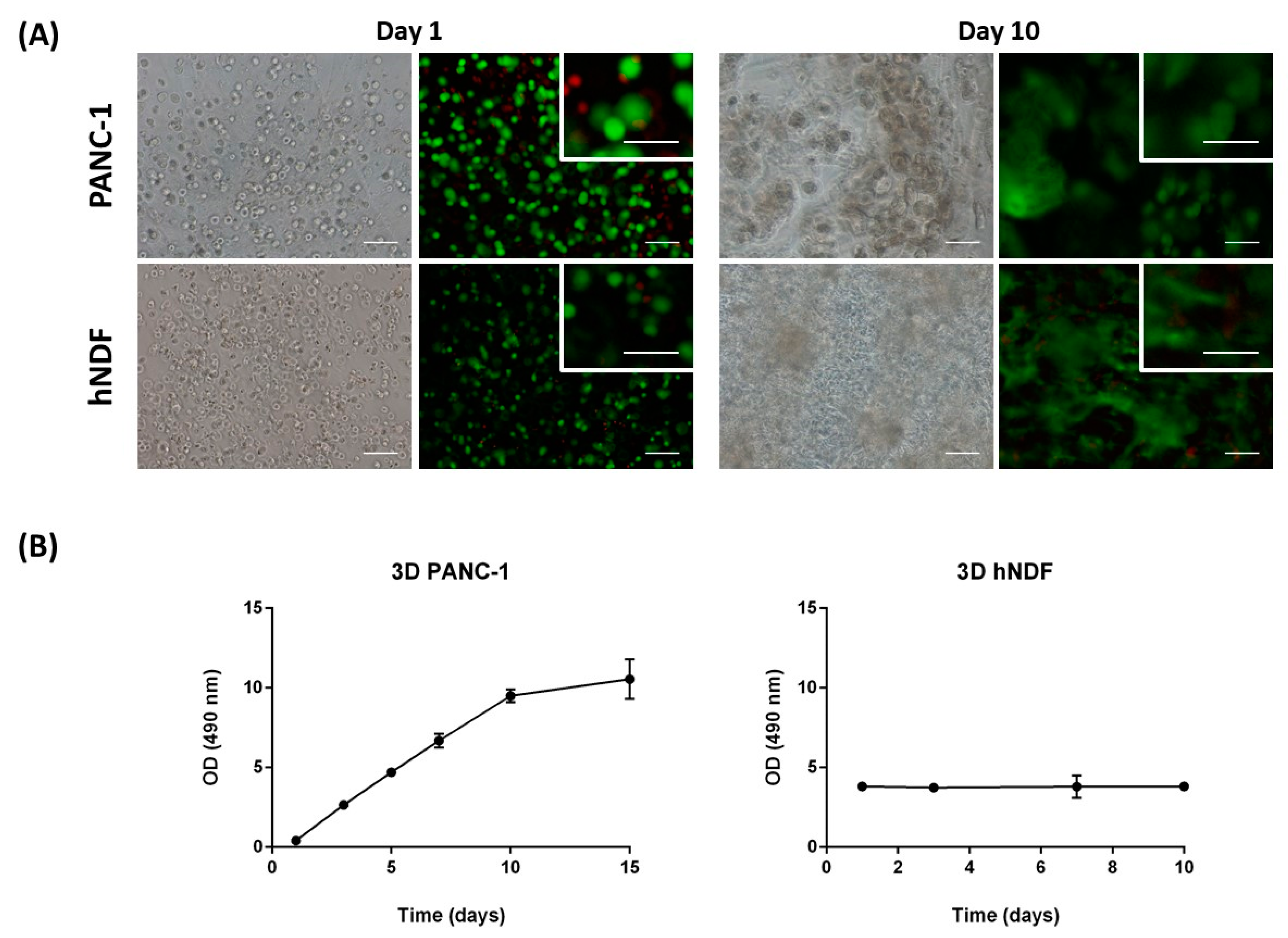
Human pancreatic ductal adenocarcinoma cells (PANC-1) were purchased from American Type Culture Collection (ATTC, CRL-1469). Primary human normal dermal fibroblasts (hNDF) were purchased from Promocell (C-12302). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, 12491-023; Termo Fisher Scientific, Waltham, MA, USA) supplemented with fetal bovine serum (FBS, S1810; Biowest), penicillin/streptomycin (L0022; Biowest, Nuaillé, France), and L-Glutamine (X0550; Biowest). The 0.05% trypsin-EDTA was from Capricorn Scientific (TRY-1B) and 1× PBS from Biowest (L0615). The self-assembling peptide RAD16-I (1% in water) is available under the commercial name PuraMatrixTM (354250; Corning, New York, NY, USA). Sucrose was purchased from Sigma (S0389, St. Louis, MO, USA). A LIVE/DEAD® Viability/Cytotoxicity Kit for mammalian cells was supplied by TermoFisher Scientific (L3224, Walthan, MA, USA). An MTS Cell Proliferation Assay Kit was purchased from abcam (ab197010). Sigma supplied the drugs Gemcitabine (G6423), 5-Fluorouracil (F6627), and 4-Methylumbelliferone (M1381). Images were taken with a Nikon Eclipse TE2000-1 epifluorescent microscope and a Leica M165C stereoscopic microscope. The statistical software used was GraphPad Prism 6.0 (San Diego, CA, USA).
3.2. Methods
3.2.1. 2D Mammalian Cell Culture
Human pancreatic ductal adenocarcinoma (PANC-1) cell line and human Normal Dermal Fibroblast (hNDF) were cultured and expanded in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% l-Glutamine in a humidified atmosphere at 37 °C and 5% CO2. Experiments were performed using cell passages between 4 and 20.
3.2.2. Cell Encapsulation into 3D SAPS
For cell encapsulation, we modified a protocol described in the previous work [
22]. The commercial peptide RAD16-I (1% in water) was prepared at a final concentration of 0.3% (
w/
v) in 10% sucrose and sonicated for 20 min at room temperature. Meanwhile, cells expanded in a 2D monolayer were trypsinized, recollected, and centrifuged (5 min at 60×
g) and then resuspended in 1–4 mL of 10% sucrose depending on pellet size. Cells were counted and centrifuged again (5 min at 60×
g). Then, cells were resuspended to 4×10
6 cells/mL in 10% sucrose. The 0.3% (
w/
v) peptide solution was mixed with an equal volume of cell suspension to obtain a final concentration of 2 × 10
6 cells/mL and 0.15% (
w/
v) RAD16-I in 10% (
w/
v) sucrose. Next, 40 µL of the mixture (80,000 cells) were loaded into wells of a 48-well plate not treated for cell culture, containing prewarmed media, which induced the spontaneous self-assembling of the peptide. The constructs were left in the flow cabinet for 20 min to let them gel. Then, the media was changed to favor the leaching of sucrose and the equilibration of cells, and the plate containing the 3D constructs was placed in the incubator (37 °C, 5% CO
2, humidified atmosphere). Cells were maintained in the incubator and the medium was changed every other day.
3.2.3. PANC-1 and hNDF Co-Culture in 3D SAPS
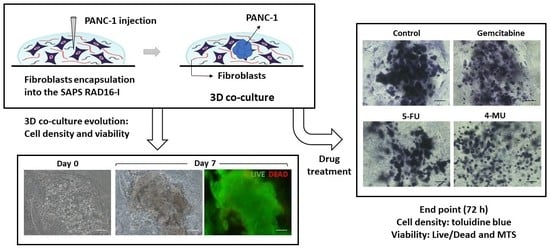
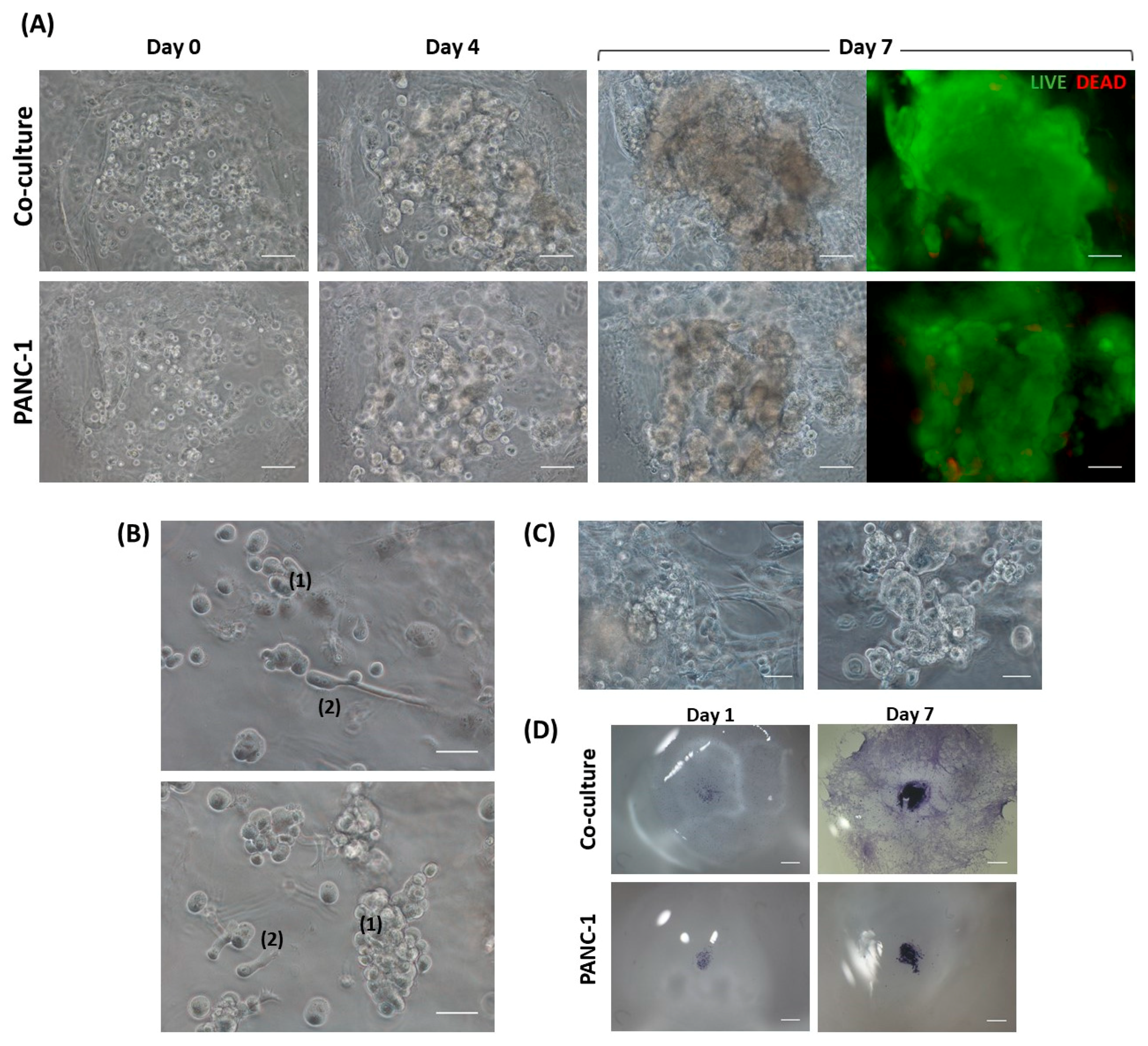
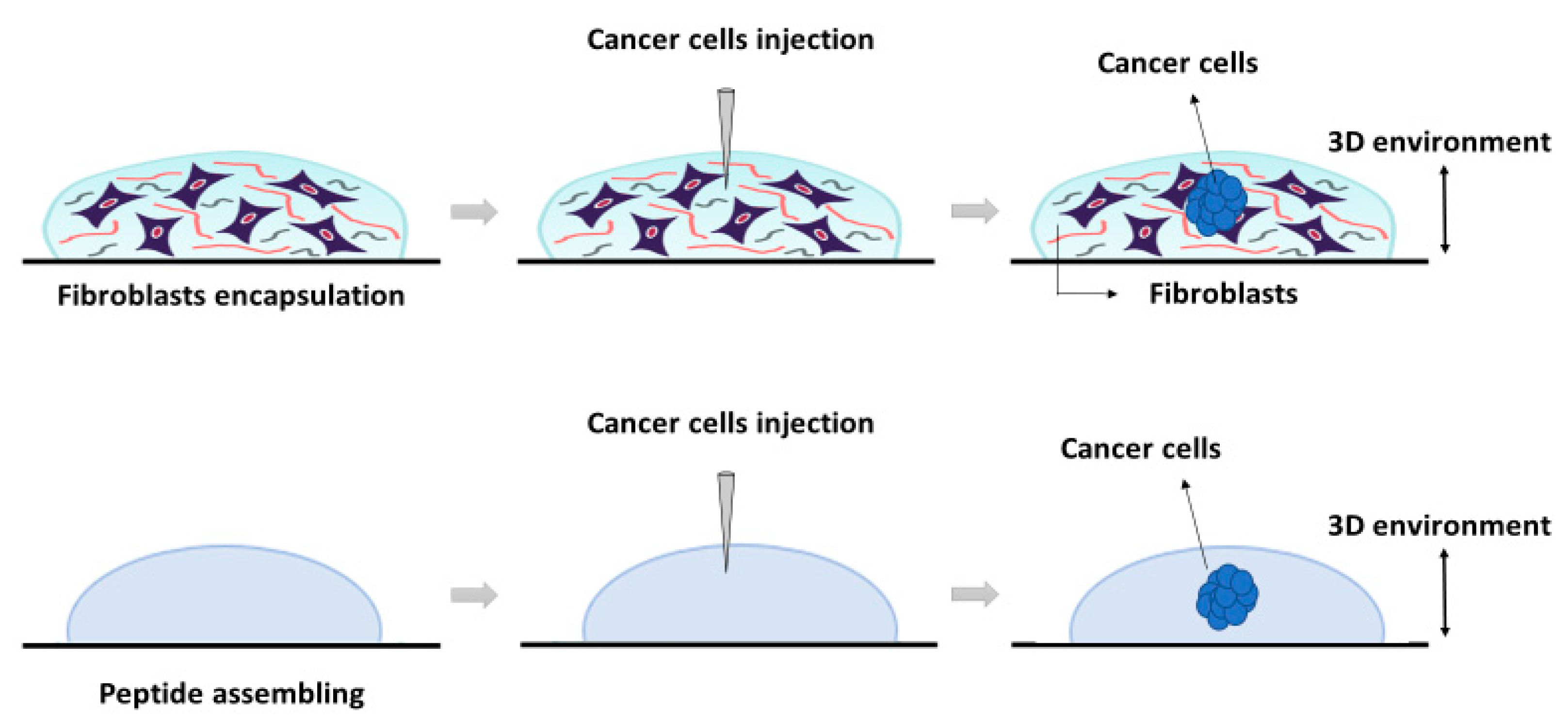
The construction of the 3D co-culture system consisted of two steps: (1) fibroblasts encapsulation within the RAD16-I peptide scaffold; and (2) cancer cells injection (
Figure 5). First, hNDF expanded in a 2D monolayer were harvested by trypsinization, centrifuged (5 min at 60×
g), and then resuspended in 1–2 mL of 10% sucrose depending on pellet size. hNDF cells were counted and centrifuged again. Then, cells were resuspended in 10% sucrose to 2.5·10
5 cells/mL. The 0.3% (
w/
v) RAD16-I peptide solution (previously sonicated) was mixed with an equal volume of cell suspension to obtain a final concentration of 1.25·10
5 cells/mL and 0.15% (
w/
v) RAD16-I in 10% sucrose. Next, 40 µL of the mixture (5000 cells) were loaded into wells of a 12- or 24-well plate. Medium was added slowly in each well, letting it slide down the wall to the gel, to induce the self-assembling of the peptide. The resulting gel was mechanically attached to the bottom of the well, which both allowed a posterior proper injection and avoided the mechanical contraction produced by fibroblasts growing in 3D gels. The 3D constructs were left in the flow cabinet for 20 min to let them gel. Then, the media was changed to favor the leaching of sucrose and the equilibration of cells, and the plate containing the hNDF 3D constructs was placed in the incubator (37 °C, 5% CO
2, humidified atmosphere). hNDF were monocultured in the 3D constructs for 3 days.
PANC-1 cells expanded in a 2D monolayer were collected by trypsinization, centrifuged, and then resuspended in 1–2 mL of 10% sucrose depending on pellet size. PANC-1 cells were counted and centrifuged again. Then, cells were resuspended in 10% sucrose to 4·106 cells/mL. The RAD16-I peptide solution (0.3% (w/v)) was mixed with an equal volume of cell suspension to obtain a final concentration of 2·106 cells/mL and 0.15% (w/v) RAD16-I in 10% sucrose. Next, 1 µL of the mixture (2000 cells) was injected, using a 10µL micropipette tip, into the 3D constructs containing hNDF cells. PANC-1 cells were also injected and monocultured in an empty gel without fibroblasts. The resulting construction consisted of a rounded gel of about 9 mm diameter, containing or not fibroblasts, with a tumor of approximately 1.5 mm diameter in the center.
3.2.4. Cell Viability
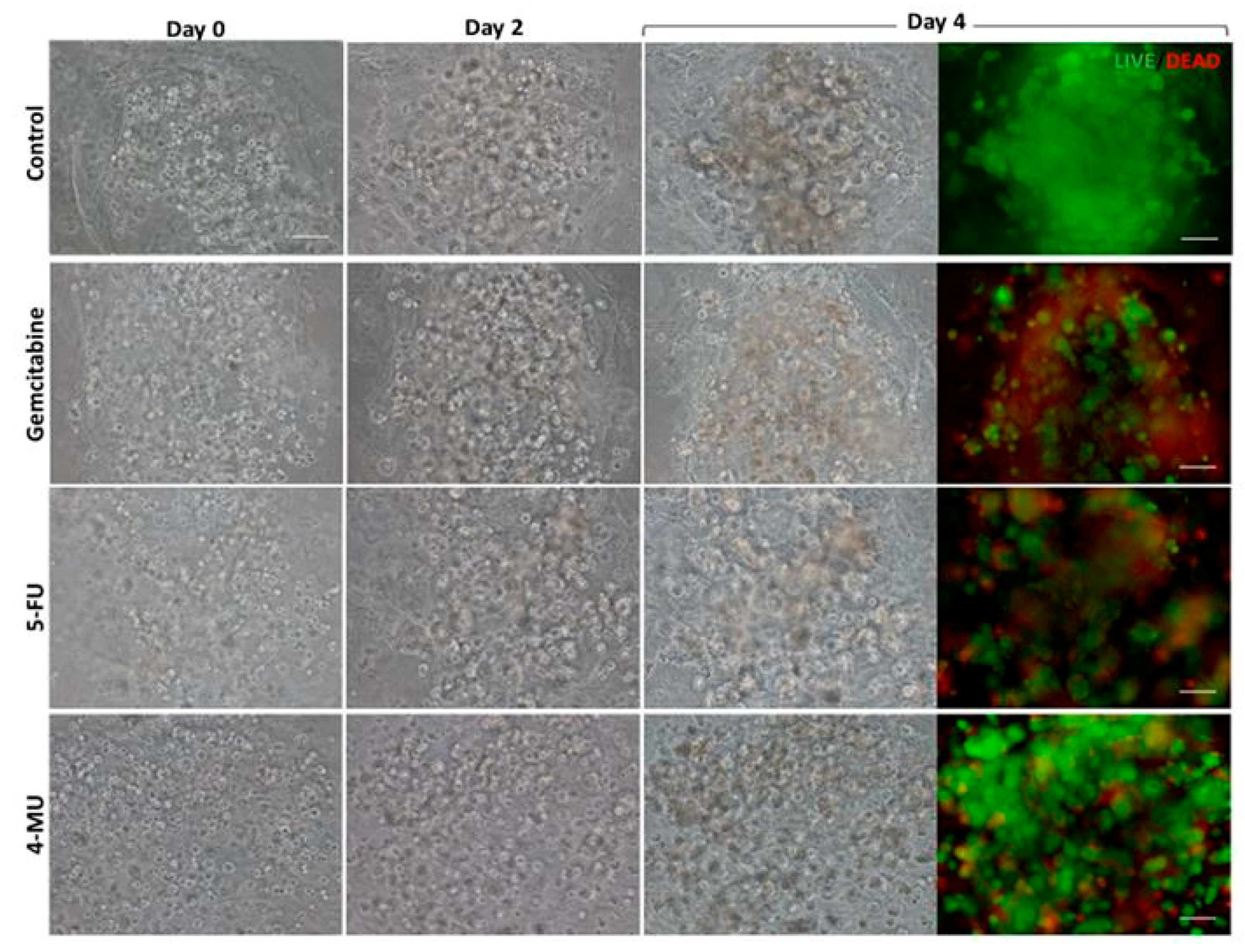
Cell viability was determined using the LIVE/DEAD® Viability/Cytotoxicity Kit for mammalian cells, which presents two components: calcein-AM and ethidium homodimer-1 (EthD-1). The biological principle of this assay is that live cells are distinguished from dead cells by the presence of intracellular esterase activity. The virtually nonfluorescent cell-permeant calcein-AM is cleaved by intracellular esterases, releasing calcein, which is highly fluorescent. The dye calcein is retained in live cells, producing an intense green fluorescence (ex/em 495/515 nm). On the other hand, EthD-1 enters only dead cells, which have damaged membranes. EthD-1 undergoes a 40-fold enhancement of fluorescence when it binds to DNA, producing a red fluorescence in dead cells (ex/em 495/635 nm). Briefly, 3D cultures were rinsed three times with 1× PBS, and then incubated for 40 min in the dark (humidified atmosphere at 37 °C and 5% CO2) with a fresh solution of 2 µM calcein and 2 µM ethidium homodimer-1 in PBS. Finally, the samples were rinsed three times with 1× PBS and then visualized under the fluorescence microscope.
3.2.5. Cell Proliferation
An MTS Assay was performed to assess cell proliferation on 3D cell culture. This assay is based on a colorimetric quantification of viable cells. Metabolically active cells reduce the MTS tetrazolium compound to generate a colored formazan product, which can be quantified by measuring the absorbance at 490–500 nm. Briefly, 3D constructs were assembled by triplicate in wells of a 48-well plate. The medium was removed, and 3D constructs were disrupted by pipetting with 1000 µL of a mixture containing MTS reagent and growth medium (20:100). Then, this mixture of cells, medium and reagent, was divided into different wells depending on cell density, and the plate was incubated in the dark for 3 h at 37 °C and 5% CO
2 in a humidified atmosphere. Next, the content of each well was collected, transferred into separate micro-centrifuge tubes, and centrifuged for 5 min at maximum speed. The aim of this step was to separate the cells and the peptide from the soluble formazan product present in the supernatant. Finally, 100 µL of the supernatant were transferred into a 96-well plate and absorbance was read at 490 nm in a microplate reader. Data was treated as follows: the optical density obtained in each well was subtracted from the blank. Then, the OD values belonging to the same replicate were summed, and finally the mean between replicates and SD were calculated. The doubling time was calculated using Equation (1).
3.2.6. Toluidine Blue Staining
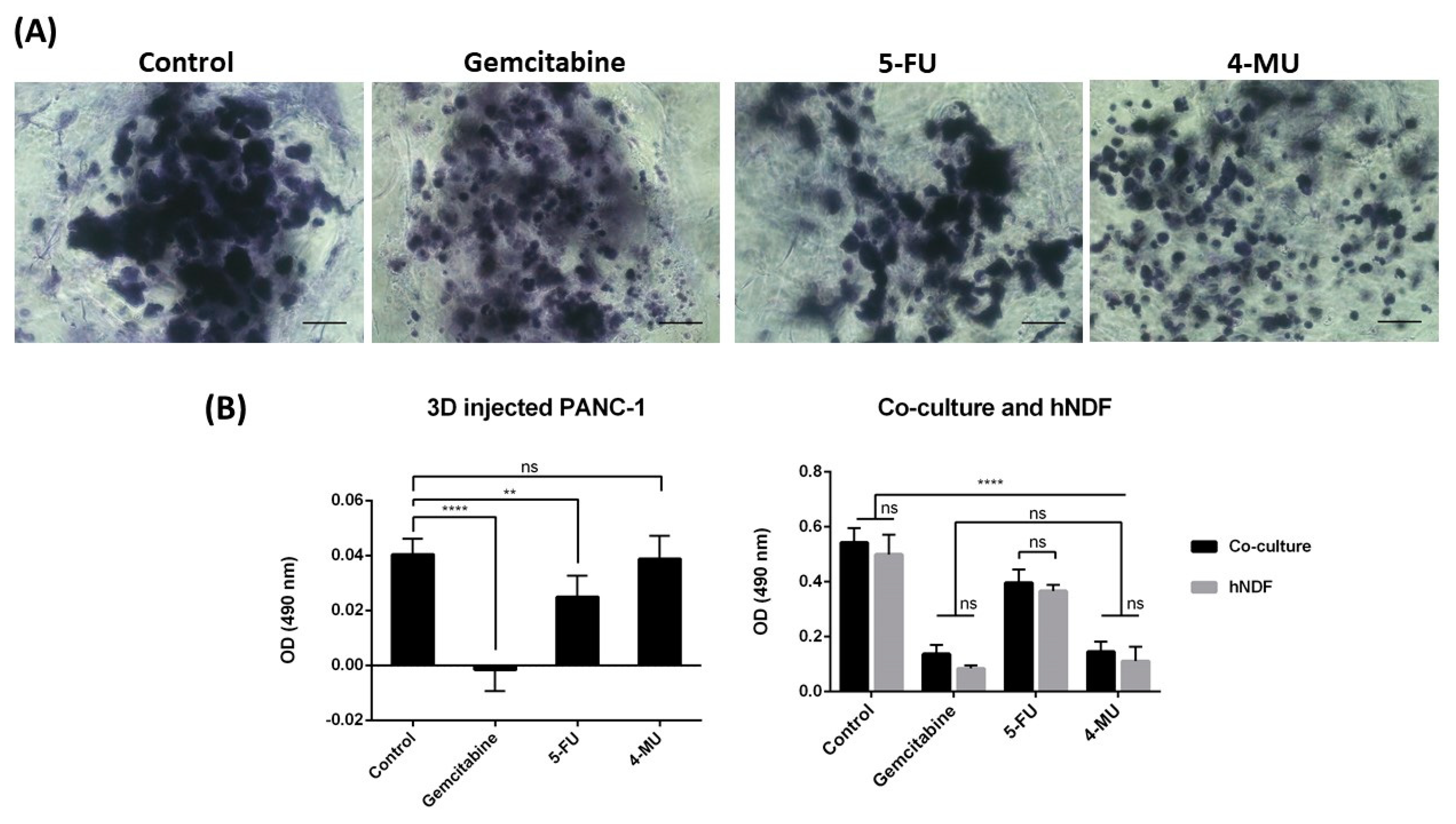
Toluidine blue (TB) is an acidophilic metachromatic dye that stains acidic tissue components (sulfates, carboxylates, and phosphate radicals). Thus, toluidine blue staining can be used as an indicator of proteoglycans (PG) production. However, in the present work, TB staining has been used to evaluate cell density in 3D cultures. For that, 3D constructs were washed with 1× PBS and fixed with 3.7% formaldehyde in PBS for 30 min. Then, cells were washed with 1× PBS and incubated with 0.05% (w/w) toluidine blue in water for 20 min. Samples were washed several times with distilled water over 24 h and visualized under contrast phase microscopy or a stereoscopic microscope.
3.2.7. Drug Incubation
The chemotherapeutic effect of different drugs was tested on 3D co-cultures, hNDF monocultures, and injected PANC-1 monocultures. The drugs and their concentrations used were: 10 µM Gemcitabine, 10 µM 5-Fluorouracil, and 1 mM 4-Methyllumbilliferone. Drugs were added 24 h after PANC-1 injection into the gel or 4 days after encapsulation in the case of hNDF monocultures. Cells were then incubated with the compounds during 72 h and assessed for viability with a Live/Dead test and an MTS assay.
3.2.8. Statistics
Data are presented as mean ± Standard Deviation. All conditions were tested by triplicate (n = 3) in three independent experiments (N = 3). One-way ANOVA was used to evaluate statistical differences between experiments. Statistical differences between groups were analyzed using Tukey’s multiple comparisons test at a significance level of 5%. The level of significance was * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, ns p > 0.05.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




