
Matured Myofibers in Bioprinted Constructs with In Vivo Vascularization and Innervation

 ,
,  , , , and
, , , and 
Abstract
:1. Introduction
2. Results
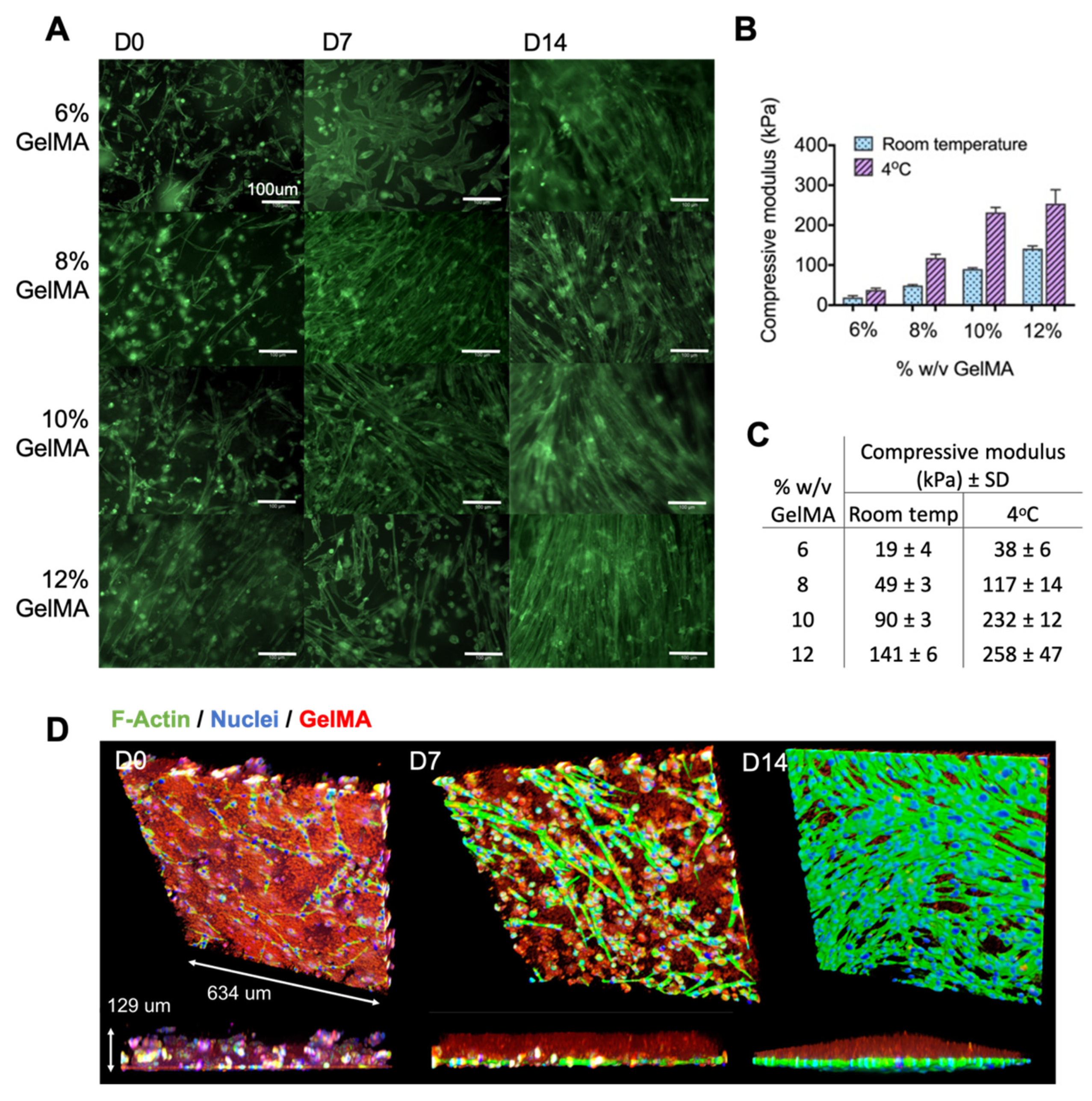
2.1. Optimizing Myoblast Culture in Cast GelMA Samples
2.2. Printing Myoblasts Encapsulated in a GelMA Bioink
2.3. Gene Expression Analysis of Bioprinted Muscle
2.4. Functional Analysis of Bioprinted Muscle
2.5. Implantation of Bioprinted Skeletal Muscle Fibers In Vivo
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. GelMA Hydrogel Synthesis
5.2. Photocuring
5.3. Rheology
5.4. Compression Testing
5.5. Primary Myoblast Cell Culture
5.6. Myoblast Encapsulation in GelMA
5.7. Bioprinting
5.8. Myoblast Differentiation
5.9. Myoblast Cell Viability
5.10. Fluorescent Staining and Imaging
5.11. RT-qPCR
5.12. Scanning Electron Microscopy (SEM) and Scanning Electron Cryomicroscopy (cryoSEM)
5.13. Calcium Imaging
5.14. In Vivo Study
5.15. Histology
5.16. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Appendix A


References
- Turner, N.J.; Badylak, S.F. Regeneration of skeletal muscle. Cell Tissue Res. 2012, 347, 759–774. [Google Scholar] [CrossRef]
- Grogan, B.F.; Hsu, J.R. Skeletal Trauma Research Consortium. Volumetric muscle loss. J. Am. Acad. Orthop. Surg. 2011, 19, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.J.; Davies, K.E. Duchenne muscular dystrophy and dystrophin: Pathogenesis and opportunities for treatment. EMBO Rep. 2004, 5, 872–876. [Google Scholar] [CrossRef]
- Cittadella Vigodarzere, G.; Mantero, S. Skeletal muscle tissue engineering: Strategies for volumetric constructs. Front. Physiol. 2014, 5, 362. [Google Scholar] [CrossRef] [Green Version]
- Ngan, C.G.Y.; Quigley, A.; Kapsa, R.M.I.; Choong, P.F.M. Engineering skeletal muscle—From two to three dimensions. J. Tissue Eng. Regen. Med. 2017, 12, e1–e6. [Google Scholar] [CrossRef]
- Jiwlawat, S.; Lynch, E.; Glaser, J.; Smit-Oistad, I.; Jeffrey, J.; Van Dyke, J.M.; Suzuki, M. Differentiation and sarcomere formation in skeletal myocytes directly prepared from human induced pluripotent stem cells using a sphere-based culture. Differentiation 2017, 96, 70–81. [Google Scholar] [CrossRef]
- Ostrovidov, S.; Ahadian, S.; Ramon-Azcon, J.; Hosseini, V.; Fujie, T.; Parthiban, S.P.; Shiku, H.; Matsue, T.; Kaji, H.; Ramalingam, M.; et al. Three-dimensional co-culture of C2C12/PC12 cells improves skeletal muscle tissue formation and function. J. Tissue Eng. Regen. Med. 2014, 11, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.S.T.; Passey, S.; Greensmith, L.; Mudera, V.; Lewis, M.P. Characterization and optimization of a simple, repeatable system for the long term in vitro culture of aligned myotubes in 3D. J Cell. Biochem. 2012, 113, 1044–1053. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Y.S.; Heinrich, M.A.; Ferrari, F.D.; Jang, H.L.; Bakht, S.M.; Alvarez, M.M.; Yang, J.; Li, Y.-C.; Santiago, G.T.; et al. Rapid Continuous Multimaterial Extrusion Bioprinting. Adv. Mater. 2017, 29, 1604630. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Seol, Y.-J.; Ko, I.K.; Kang, H.-W.; Lee, Y.K.; Yoo, J.J.; Atala, A.; Lee, S.J. 3D Bioprinted Human Skeletal Muscle Constructs for Muscle Function Restoration. Sci. Rep. 2018, 8, 12307. [Google Scholar] [CrossRef] [Green Version]
- Billiet, T.; Gevaert, E.; De Schryver, T.; Cornelissen, M.; Dubruel, P. The 3D printing of gelatin methacrylamide cell-laden tissue-engineered constructs with high cell viability. Biomaterials 2014, 35, 49–62. [Google Scholar] [CrossRef]
- Duchi, S.; Onofrillo, C.; O’Connell, C.D.; Blanchard, R.; Augustine, C.; Quigley, A.F.; Kapsa, R.M.I.; Pivonka, P.; Wallace, G.; Di Bella, C.; et al. Handheld Co-Axial Bioprinting: Application to in situ surgical cartilage repair. Sci. Rep. Nat. 2017, 7, 5837. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Heinrich, M.A.; Zhou, Y.; Akpek, A.; Hu, N.; Liu, X.; Guan, X.; Zhong, Z.; Jin, X.; Khademhosseini, A.; et al. Extrusion Bioprinting of Shear-Thinning Gelatin Methacryloyl Bioinks. Adv. Healthc. Mater. 2017, 6, 1601451. [Google Scholar] [CrossRef]
- Shi, X.; Ostrovidov, S.; Zhao, Y.; Liang, X.; Kasuya, M.; Kurihara, K.; Nakajima, K.; Bae, H.; Wu, H.; Khademhosseini, A. Microfluidic Spinning of Cell-Responsive Grooved Microfibers. Adv. Funct. Mater. 2015, 25, 2250–2259. [Google Scholar] [CrossRef]
- Aubin, H.; Nichol, J.W.; Hutson, C.B.; Bae, H.; Sieminski, A.L.; Cropek, D.M.; Akhyari, P.; Khademhosseini, A. Directed 3D cell alignment and elongation in microengineered hydrogels. Biomaterials 2010, 31, 6941–6951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, V.; Ahadian, S.; Ostrovidov, S.; Camci-Unal, G.; Chen, S.; Kaji, H.; Ramalingam, M.; Khademhosseini, A. Engineered Contractile Skeletal Muscle Tissue on a Microgrooved Methacrylated Gelatin Substrate. Tissue Eng. Part A 2012, 18, 2453–2465. [Google Scholar] [CrossRef] [Green Version]
- Ostrovidovm, S.; Shi, X.; Zhang, L.; Liang, X.; Kim, S.B.; Fujie, T.; Ramalingam, M.; Chen, M.; Nakajima, K.; Al-Hazmi, F.; et al. Myotube formation on gelatin nanofibers—Multi-walled carbon nanotubes hybrid scaffolds. Biomaterials 2014, 35, 6268–6277. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.-W.; Lee, S.J.; Ko, I.K.; Kengla, C.; Yoo, J.J.; Atala, A. A 3D Bioprinting System to Produce Human-Scale Tissue Constructs with Structural Integrity. Nat. Biotechnol. 2016, 34, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Ursu, D.C.; Urbanchek, M.G.; Nedic, A.; Cederna, P.S.; Gillespie, R.B. In vivo characterization of regenerative peripheral nerve interface function. J. Neural Eng. 2016, 13, 026012. [Google Scholar] [CrossRef] [PubMed]
- Mian, R.; Morrison, W.A.; Hurley, J.V.; Penington, A.; Romeo, R.; Tanaka, Y.; Knight, K.R. Formation of new tissue from an arteriovenous loop in the absence of added extracellular matrix. Tissue Eng. 2000, 6, 595–603. [Google Scholar] [CrossRef]
- Boos, A.M.; Loew, J.S.; Weigand, A.; Deschler, G.; Klumpp, D.; Arkudas, A.; Bleiziffer, O.; Gulle, H.; Kneser, U.; Horch, R.E.; et al. Engineering axially vascularized bone in the sheep arteriovenous-loop model. J. Tissue Eng. Regen. Med. 2012, 22, 654–664. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tsutsumi, A.; Crowe, D.M.; Tajima, S.; Morrison, W.A. Generation of an autologous tissue (matrix) flap by combining an arteriovenous shunt loop with artificial skin in rats: Preliminary report. Br. J. Plast. Surg. 2000, 53, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Messina, A.; Bortolotto, S.K.; Cassell, O.C.S.; Kelly, J.; Abberton, K.M.; Morrison, W.A. Generation of a vascularized organoid using skeletal muscle as the inductive source. FASEB J. Fed. Am. Soc. Exp. Biol. 2005, 19, 1570–1572. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, I.; Seol, Y.-J.; Ko, I.K.; Yoo, J.J.; Atala, A.; Lee, S.J. Neural Cell Integration into 3D Bioprinted Skeletal Muscle Constructs Accelerates Restoration of Muscle Function. Nat. Commun. 2020, 11, 1025. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building Muscle: Molecular Regulation of Myogenesis. Biology 2012, 4, a008342. [Google Scholar] [CrossRef]
- Schiaffino, S.; Rossi, A.C.; Smerdu, V.; Leinwand, L.A.; Reggiani, C. Developmental myosins: Expression patterns and functional significance. Skelet. Muscle 2015, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Hernández, J.M.; García-González, E.G.; Brun, C.E.; Rudnicki, M.A. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 2017, 72, 10–18. [Google Scholar] [CrossRef]
- Grifone, R.; Demignon, J.; Houbron, C.; Souil, E.; Niro, C.; Seller, M.J.; Hamard, G.; Maire, P. Six1 and Six4 homeoproteins are required for Pax3 and Mrf expression during myogenesis in the mouse embryo. Development 2005, 132, 2235–2249. [Google Scholar] [CrossRef] [Green Version]
- Capote, J.; Bolaños, P.; Schuhmeier, R.P.; Melzer, W.; Caputo, C. Calcium transients in developing mouse skeletal muscle fibres. J. Physiol. 2005, 564, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Sciancalepore, M.; Afzalov, R.; Buzzin, V.; Jurdana, M.; Lorenzon, P.; Ruzzier, F. Intrinsic ionic conductances mediate the spontaneous electrical activity of cultured mouse myotubes. Biochim. Biophys. Acta 2005, 1720, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Cognard, C.; Constantin, B.; Rivet-Bastide, M.; Imbert, N.; Besse, C.; Raymond, G. Appearance and evolution of calcium currents and contraction during the early post-fusional stages of rat skeletal muscle cells developing in primary culture. Development 1993, 117, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, M.; Wang, J.; Zhang, W.; Lu, W.; Gao, Y.; Zhang, B.; Guo, Y. Development of a Photo-Crosslinking, Biodegradable GelMA/PEGDA Hydrogel for Guided Bone Regeneration Materials. Materials 2018, 11, 1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engler, A.J.; Griffin, M.A.; Sen, S.; Bönnemann, C.G.; Sweeney, H.L.; Discher, D.E. Myotubes differentiate optimally on substrates with tissue-like stiffness. J. Cell Biol. 2004, 166, 877–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, M.A.; Sen, S.; Sweeney, H.L.; Discher, D.E. Adhesion-contractile balance in myocyte differentiation. J. Cell Sci. 2004, 117, 5855–5863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Krishnan, U.M.; Sethuraman, S. Development of biomaterial scaffold for nerve tissue engineering: Biomaterial mediated neural regeneration. J. Biomed. Sci. 2009, 16, 108. [Google Scholar]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Aparnathi, M.K.; Patel, J.S. Biodegradable Gelatin Methacrylate Gel as a Potential Scaffold for Bone Tissue Engineering of Canine Adipose-Derived Stem Cells. J. Stem Cells 2016, 11, 111–119. [Google Scholar]
- Zhao, X.; Sun, X.; Yildirimer, L.; Lang, Q.; Lin, Z.Y.; Zheng, R.; Zhang, Y.; Cui, W.; Annabi, N.; Khademhosseini, A. Cell infiltrative hydrogel fibrous scaffolds for accelerated wound healing. Acta Biomater. 2017, 49, 66–77. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, C.D.; Bella, C.D.; Thompson, F.; Augustine, C.; Beirne, S.; Cornock, R.; Richards, C.J.; Chung, J.; Gambhir, S.; Yue, Z.; et al. Development of the Biopen: A handheld device for surgical printing of adipose stem cells at a chondral wound site. Biofabrication 2016, 8, 015019. [Google Scholar] [CrossRef]
- Loessner, D.; Meinert, C.; Kaemmerer, E.; Martine, L.C.; Yue, K.; Levett, P.A.; Klein, T.J.; Melchels, P.; Khademhosseini, A. Functionalization, preparation and use of cell-laden gelatin methacryloyl-based hydrogels as modular tissue culture platforms. Nat. Protoc. 2016, 11, 727–746. [Google Scholar] [CrossRef] [Green Version]
- Todaro, M.; Quigley, A.; Kita, M.; Chin, J.; Lowes, K.; Kornberg, A.J.; Cook, M.J.; Kapsa, R. Effective detection of corrected dystrophin loci in mdx mouse myogenic precursors. Hum. Mutat. 2007, 28, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Starly, B.; Daly, A.C.; Burdick, J.A.; Groll, J.; Skeldon, G.; Shu, W.; Sakai, Y.; Shinohara, M.; Nishikawa, M.; et al. The Bioprinting Roadmap. Biofabrication 2020, 12, 022002. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C-T method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Jaimovich, E.; Reyes, R.; Liberona, J.L.; Powell, J.A. IP 3receptors, IP 3transients, and nucleus-associated Ca 2+signals in cultured skeletal muscle. Am. J. Physiol. Cell Physiol. 2000, 278, C998–C1010. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, S.L.; Crook, J.M.; Morrison, W.A.; Newcomb, A.E. Toward clinical application of stem cells for cardiac regeneration. Heart Lung Circ. 2011, 20, 173–179. [Google Scholar] [CrossRef]
- Ngan, C.G.Y.; O’Connell, C.D.; Blanchard, R.; Boyd-Moss, M.; Williams, R.J.; Bourke, J.; Quigley, A.; McKelvie, P.; Kapsa, R.M.I.; Choong, P.F.M. Optimising the biocompatibility of 3D printed photopolymer constructs in vitro and in vivo. Biomed. Mater. 2019, 14, 035007. [Google Scholar] [CrossRef]
- O’Connell, C.D.; Zhang, B.; Onofrillo, C.; Duchi, S.; Blanchard, R.; Quigley, A.; Bourke, J.; Gambhir, S.; Kapsa, R.; Bella, C.D.; et al. Tailoring the mechanical properties of gelatin methacryloyl hydrogels through manipulation of the photocrosslinking conditions. Soft Matter 2018, 14, 2142–2151. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Forward Primer | Reverse Primer |
|---|---|---|
| Myogenin (MYOG) | 5′-GCGCCATCCAGTACATTGAGC-3′ | 5′-ACGATGGACGTAAGGGAGTGC-3′ |
| Myogenic factor 6 (MYF6) | 5′CGGAGTGCCATCAGCTACATTG-3′ | 5′-TCCACGTTTGCTCCTCCTTCC-3′ |
| Homeobox protein SIX4 (SIX4) | 5′-TTC AAG GAG AAG TCG CGC AAC-3′ | 5′-ACT GGG GTT GCC ATC CGA TTC-3′ |
| Myosin heavy chain 1 (MYH1) | 5′-GGCACTGTGGACTACAACATCG-3′ | 5′-TTT CTT TCC ACC ACC GCC ACC-3′ |
| Myosin heavy chain 8 (MYH8) | 5′-CTACCAAAGGCAAGGCCGAG-3′ | 5′-ATCTGCTTCAGCACTAGCGTATG-3′ |
| Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) | 5′-ACAACTTTGGCATTGTGGAAGGG-3′ | 5′-TACTTGGCAGGTTTCTCCAGGC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngan, C.G.Y.; Quigley, A.; Williams, R.J.; O’Connell, C.D.; Blanchard, R.; Boyd-Moss, M.; Aumann, T.D.; McKelvie, P.; Wallace, G.G.; Choong, P.F.M.; et al. Matured Myofibers in Bioprinted Constructs with In Vivo Vascularization and Innervation. Gels 2021, 7, 171. https://doi.org/10.3390/gels7040171
Ngan CGY, Quigley A, Williams RJ, O’Connell CD, Blanchard R, Boyd-Moss M, Aumann TD, McKelvie P, Wallace GG, Choong PFM, et al. Matured Myofibers in Bioprinted Constructs with In Vivo Vascularization and Innervation. Gels. 2021; 7(4):171. https://doi.org/10.3390/gels7040171
Chicago/Turabian StyleNgan, Catherine G. Y., Anita Quigley, Richard J. Williams, Cathal D. O’Connell, Romane Blanchard, Mitchell Boyd-Moss, Tim D. Aumann, Penny McKelvie, Gordon G. Wallace, Peter F. M. Choong, and et al. 2021. "Matured Myofibers in Bioprinted Constructs with In Vivo Vascularization and Innervation" Gels 7, no. 4: 171. https://doi.org/10.3390/gels7040171
APA StyleNgan, C. G. Y., Quigley, A., Williams, R. J., O’Connell, C. D., Blanchard, R., Boyd-Moss, M., Aumann, T. D., McKelvie, P., Wallace, G. G., Choong, P. F. M., & Kapsa, R. M. I. (2021). Matured Myofibers in Bioprinted Constructs with In Vivo Vascularization and Innervation. Gels, 7(4), 171. https://doi.org/10.3390/gels7040171