

Self-Healing and Super-Elastomeric PolyMEA-co-SMA Nanocomposites Crosslinked by Clay Platelets

 ,
,
Abstract
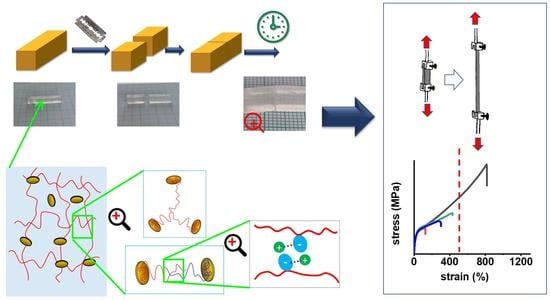
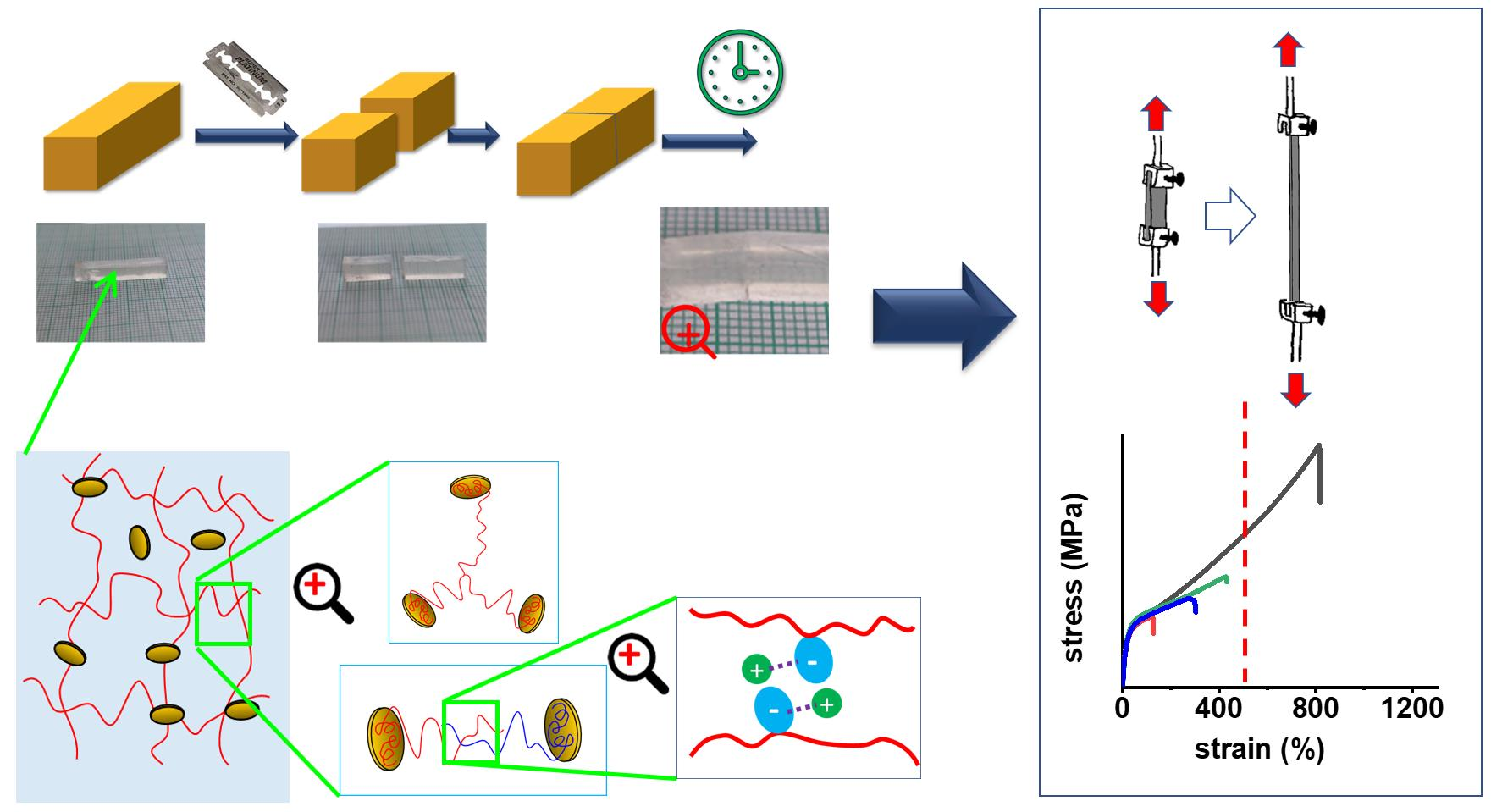
1. Introduction
2. Results and Discussion
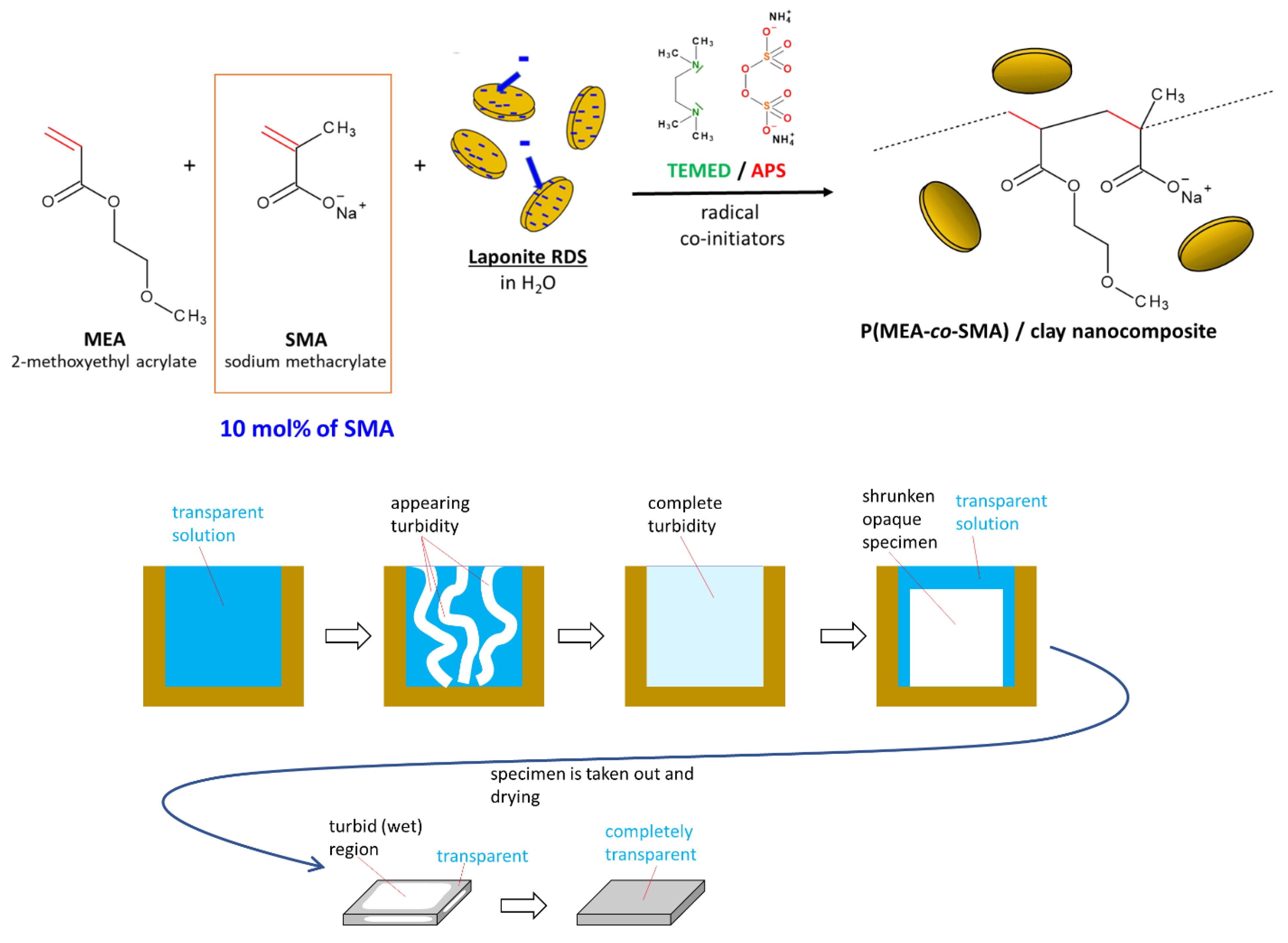
2.1. Synthesis of the Super-Elastomeric Nanocomposites
2.1.1. General Properties, Hydrophobicity vs. Hydrophilicity
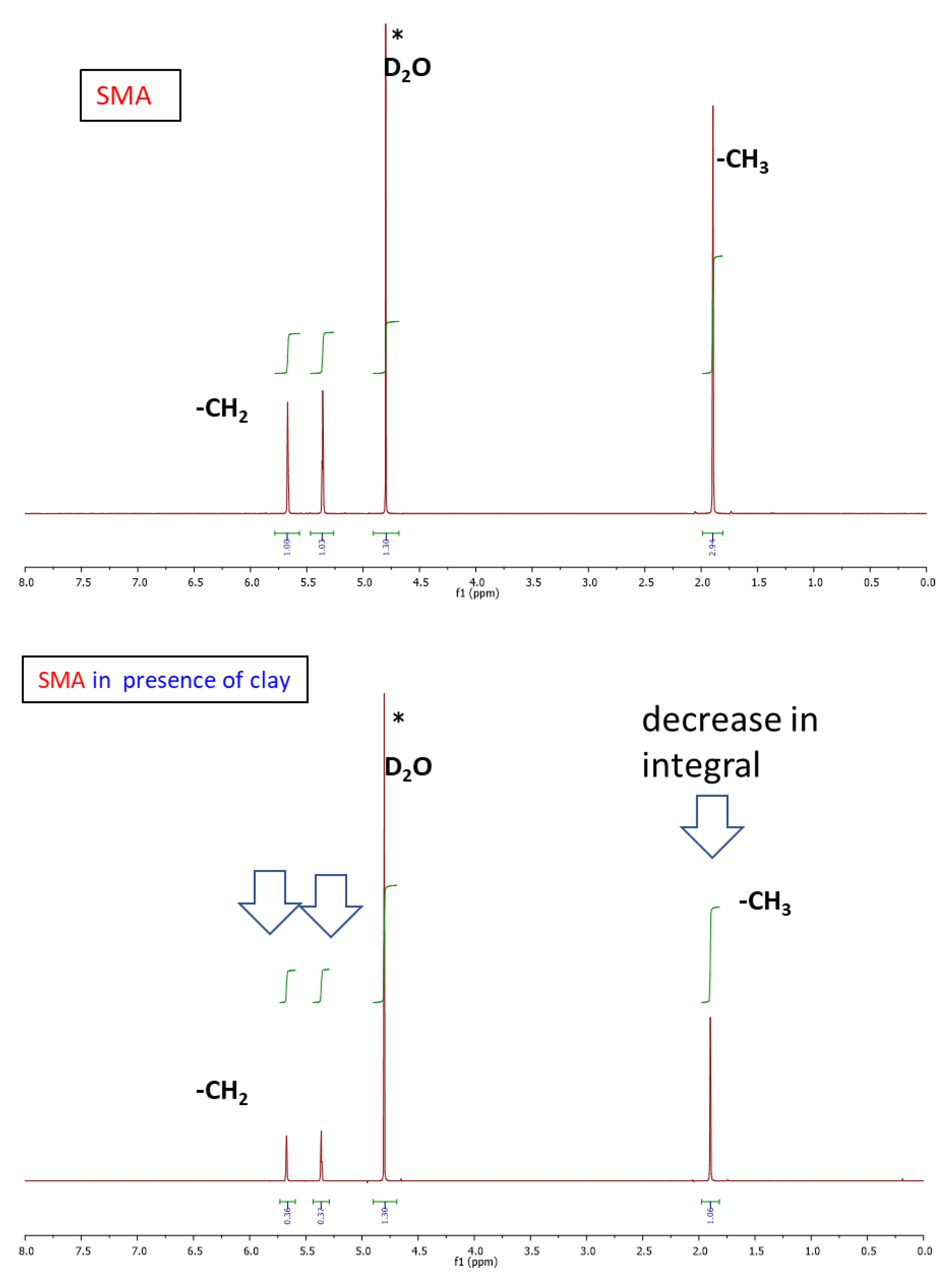
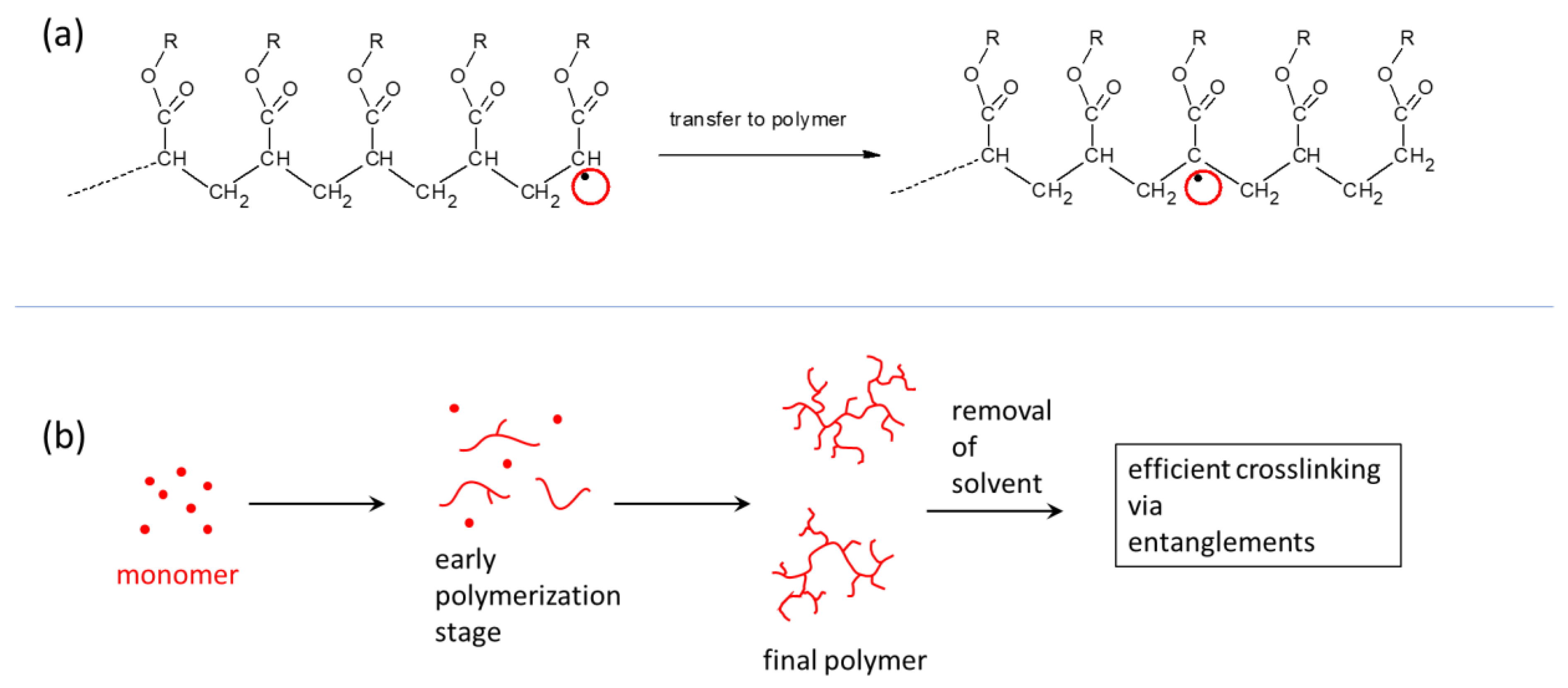
2.1.2. Structure of Crosslinking in the Nanocomposites
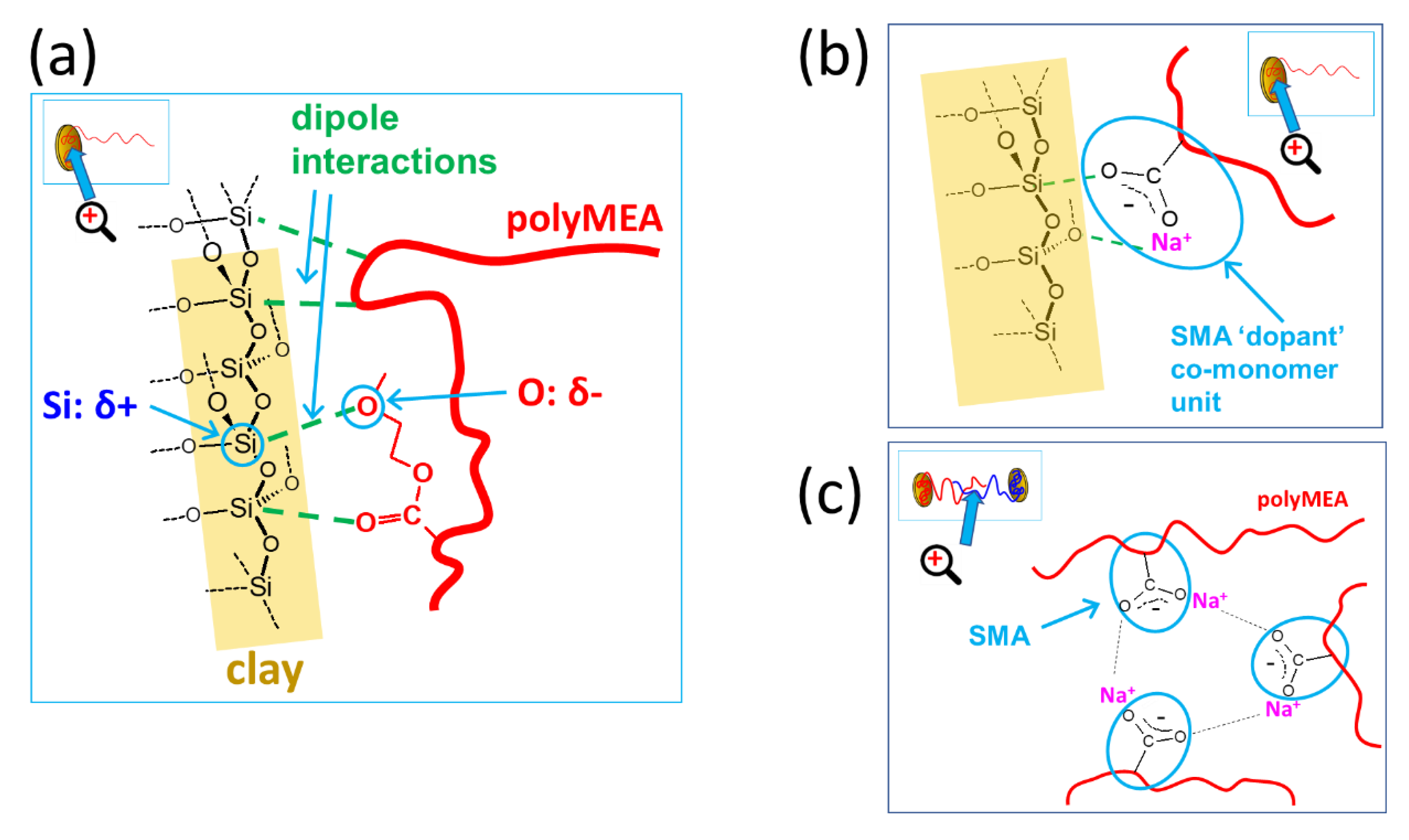
Interactions in the Nanocomposites
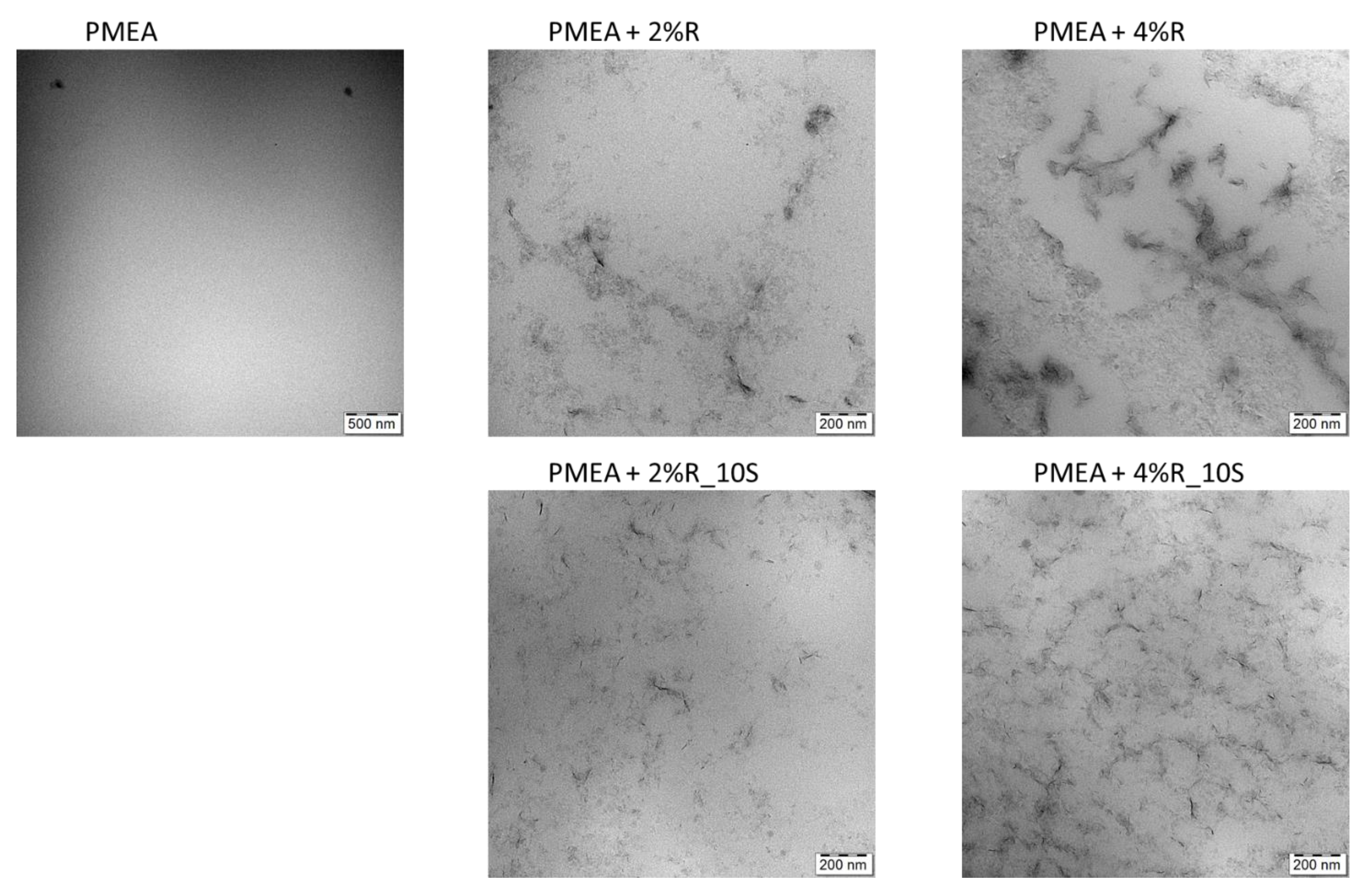
2.1.3. Morphology (TEM)
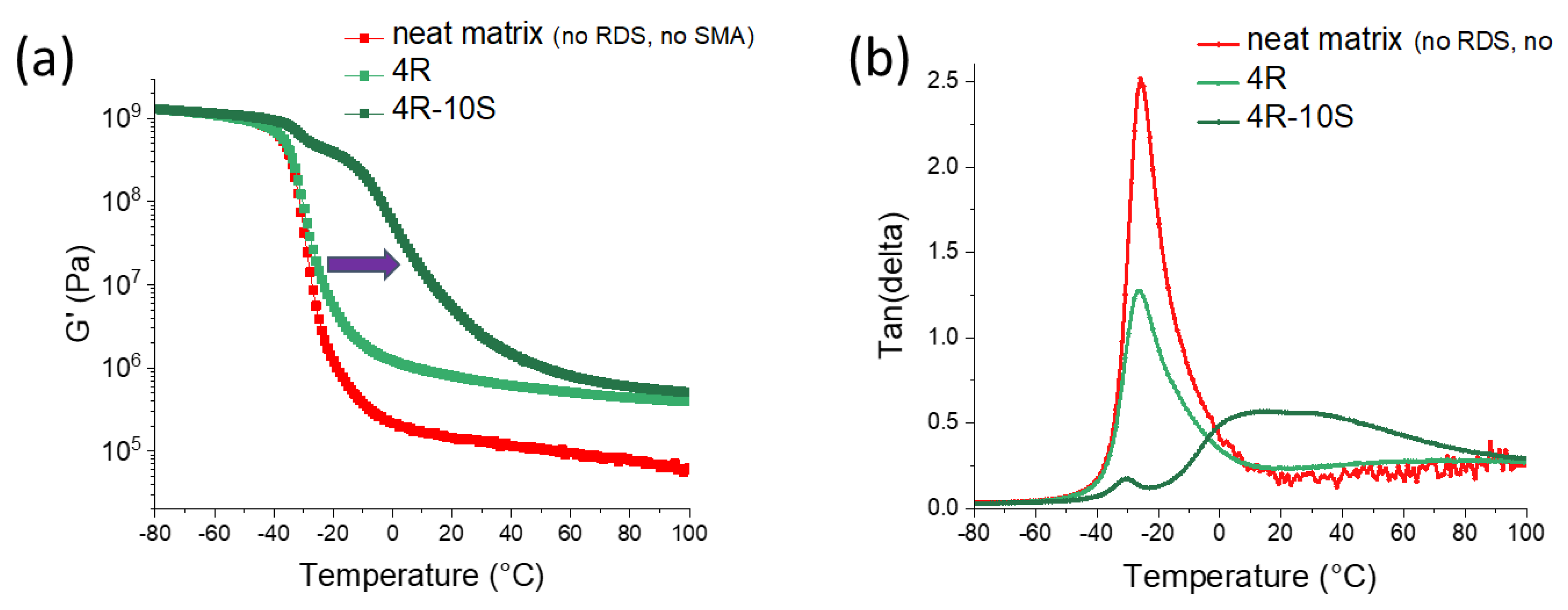
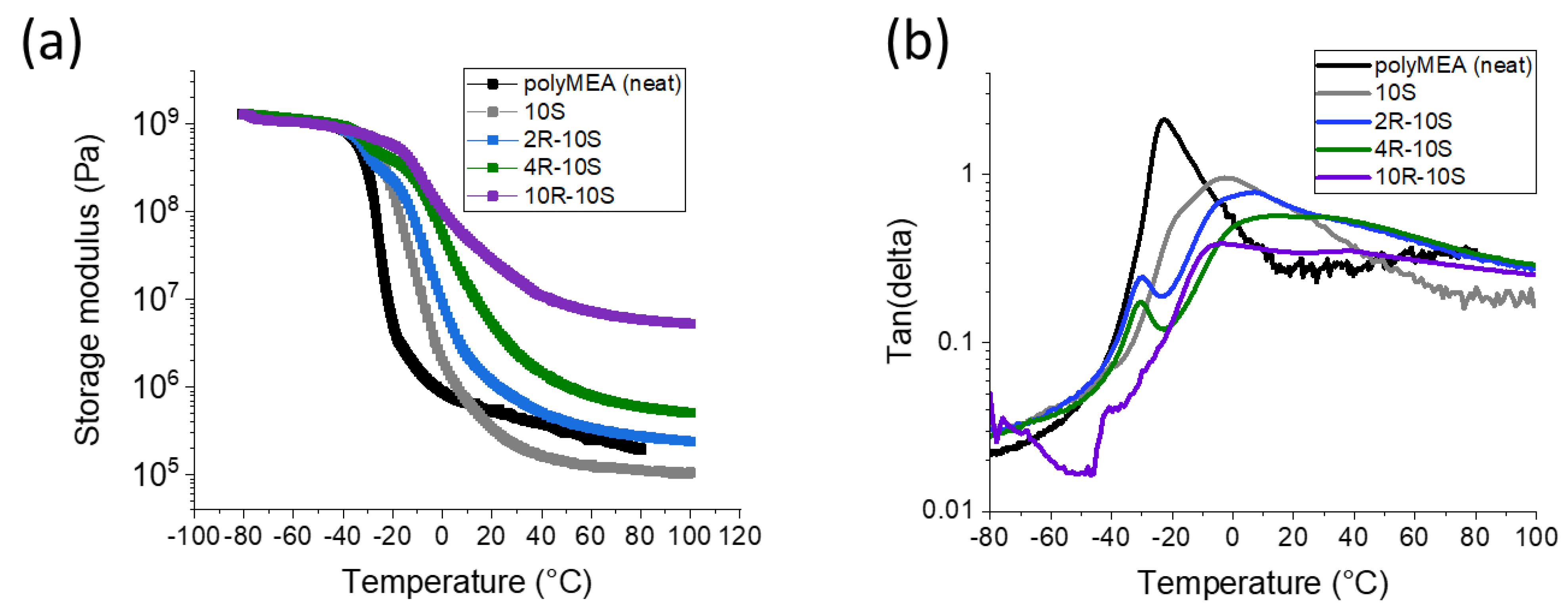
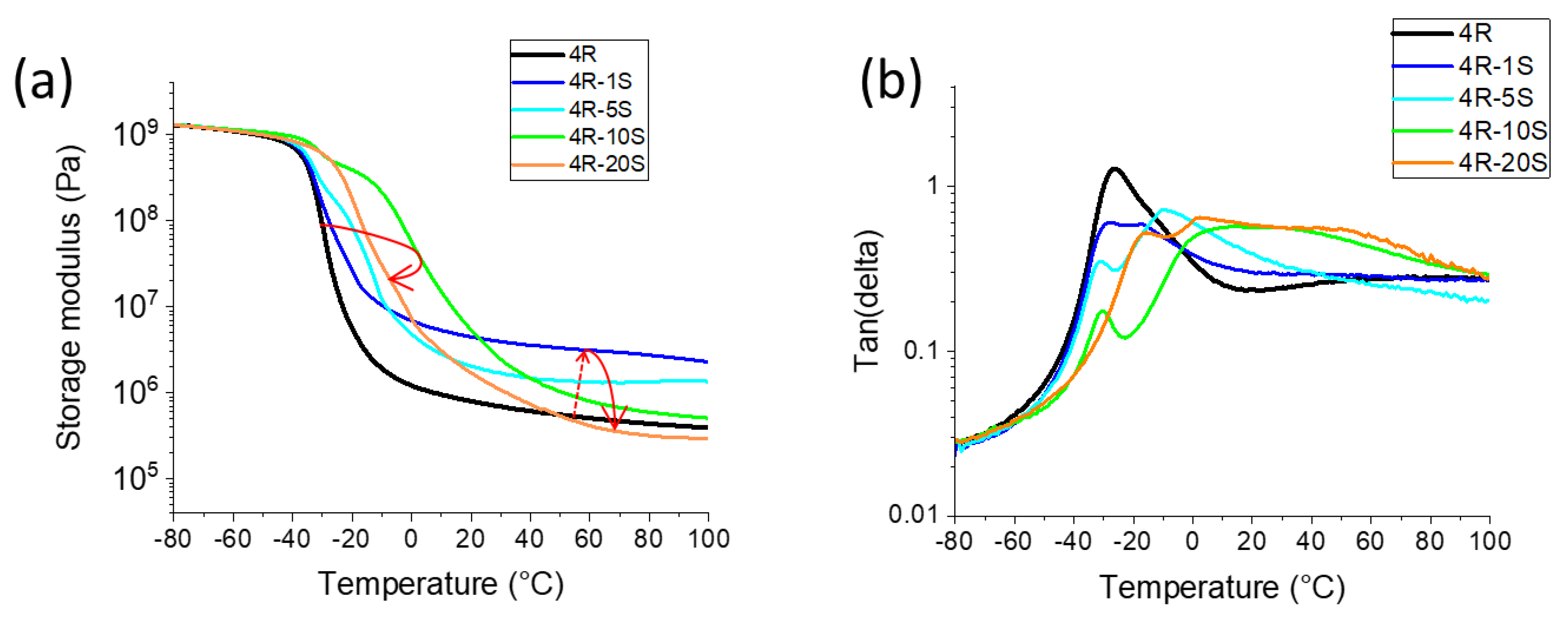
2.2. Thermo-Mechanical Properties (DMTA)
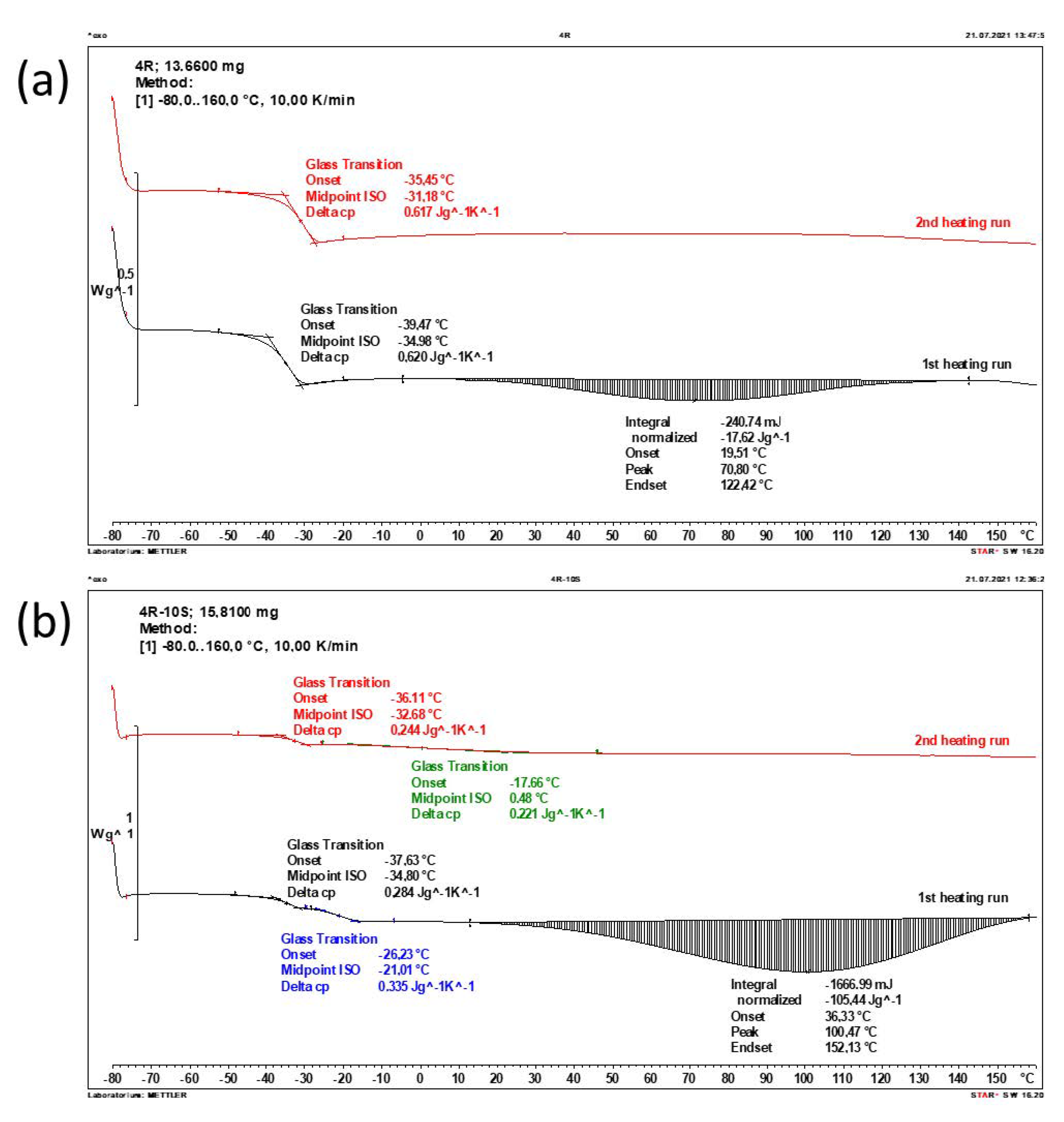
2.3. Phase Transition Behaviour and Thermal Properties (DSC)
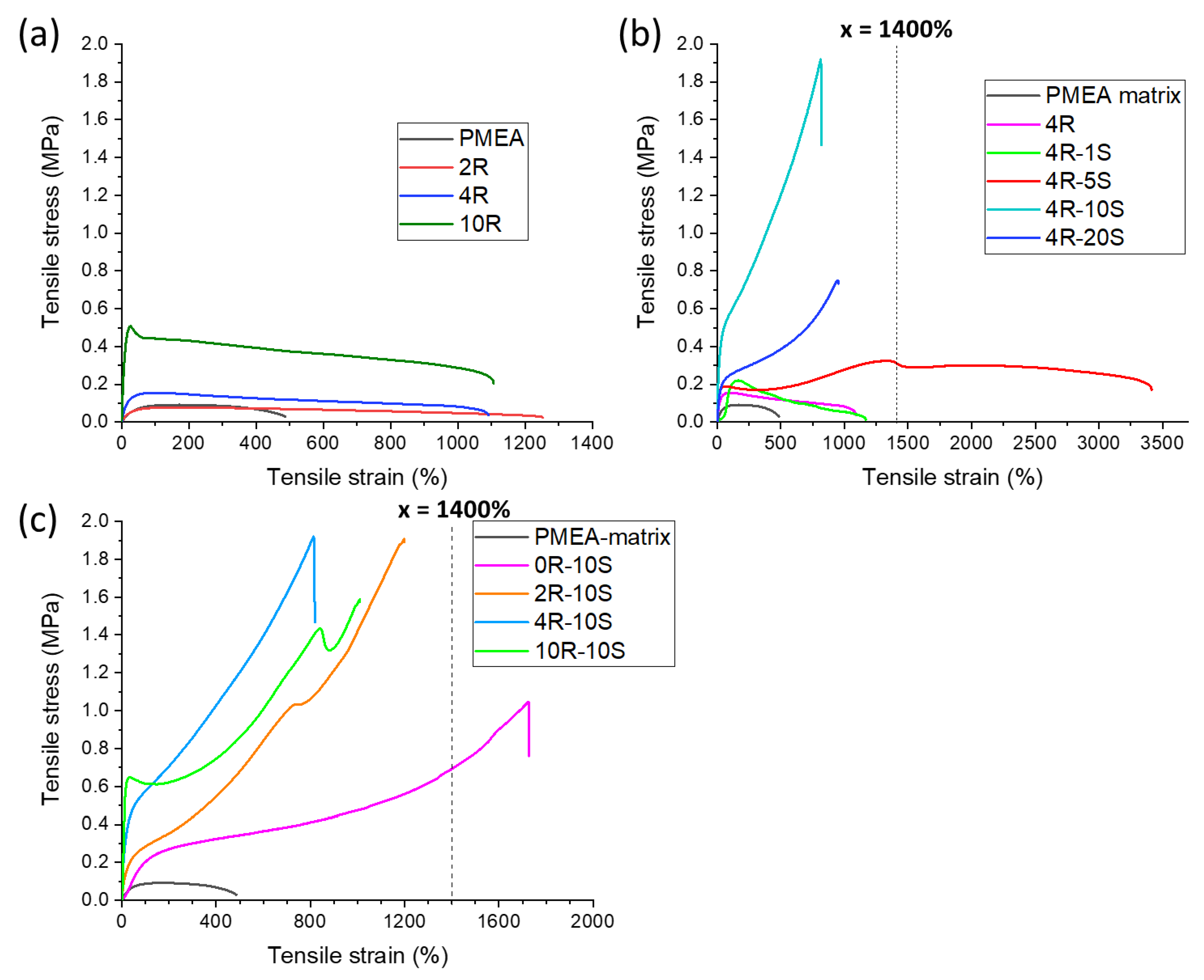
2.4. Tensile Properties: Simple Tests
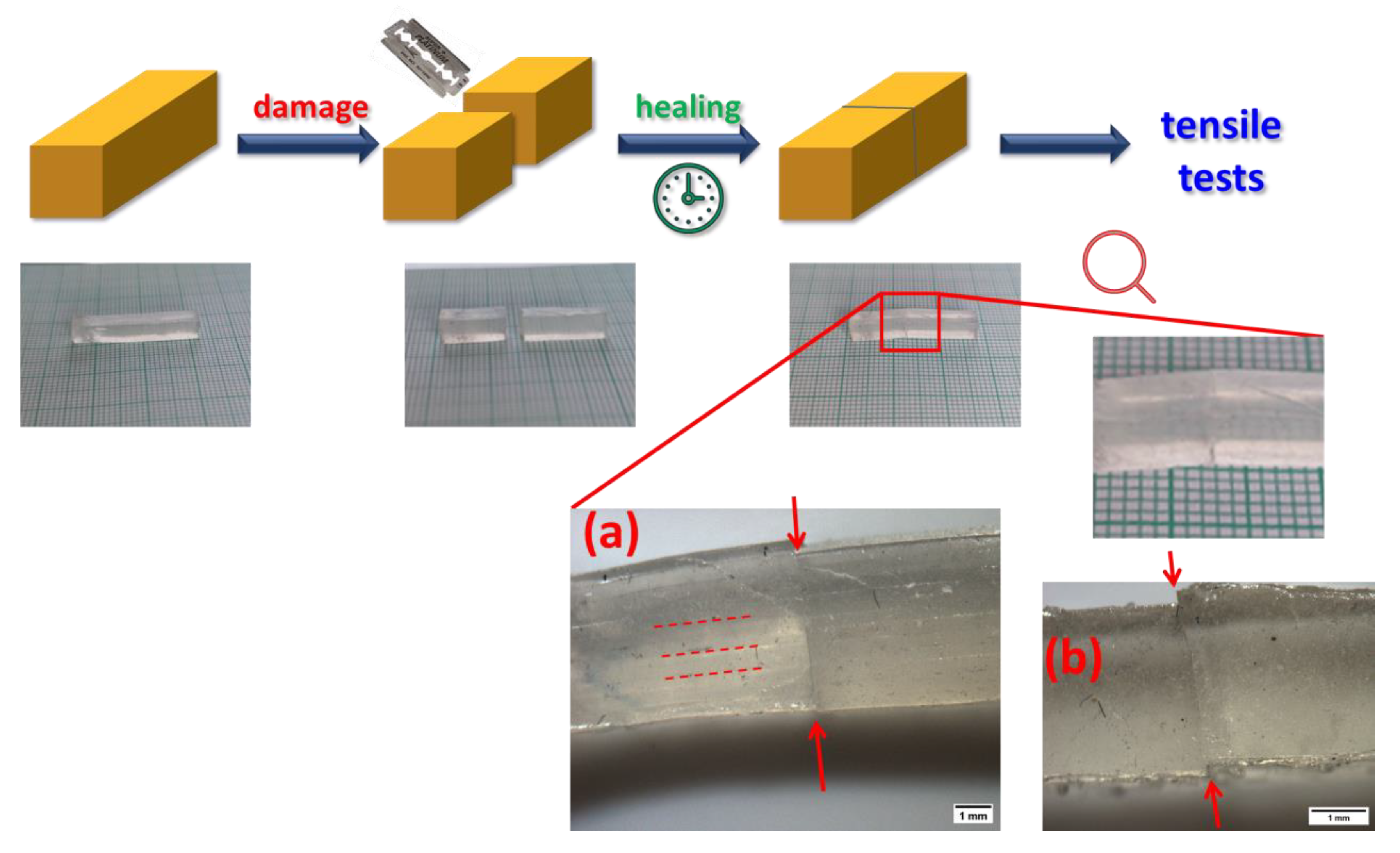
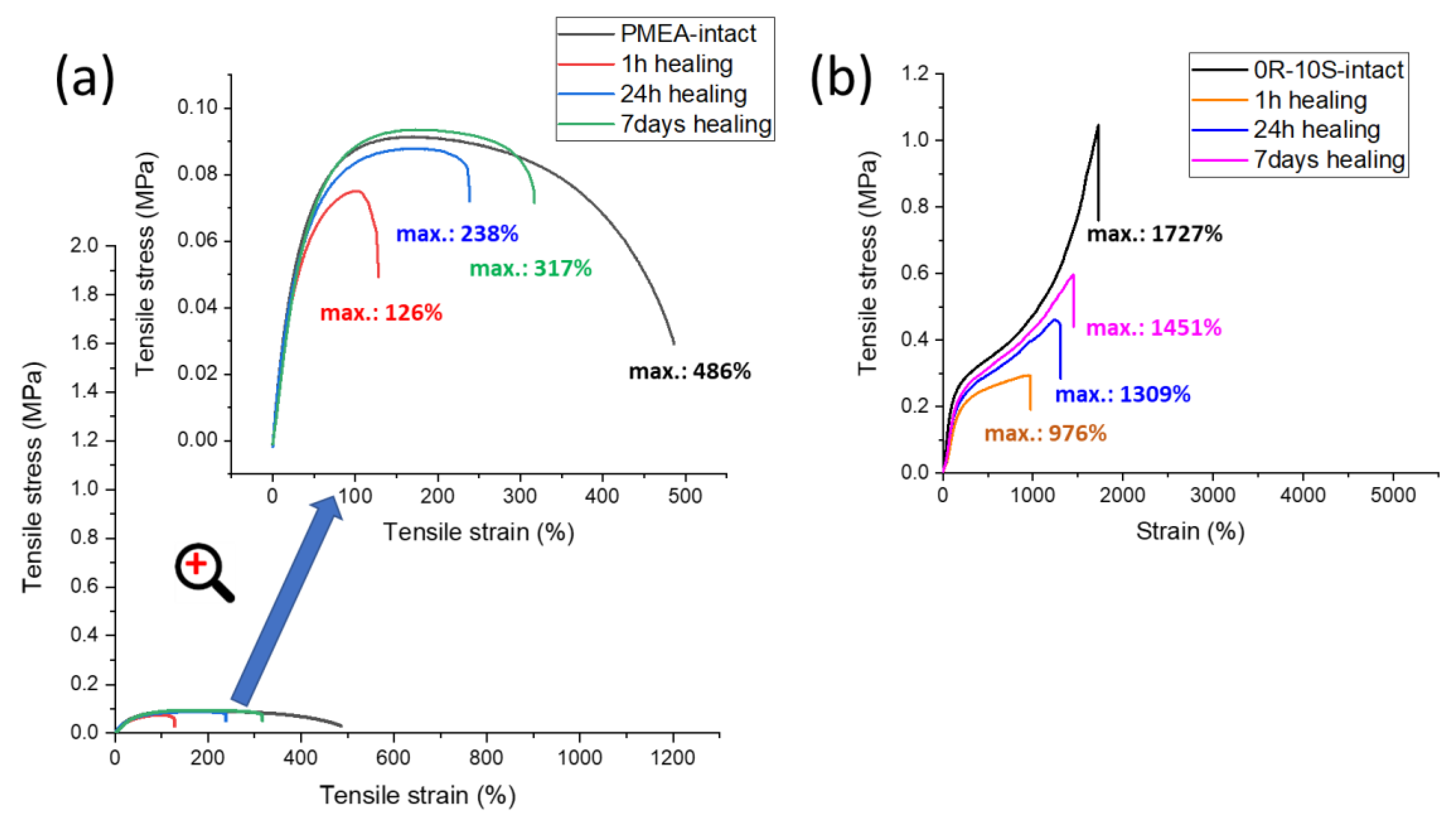
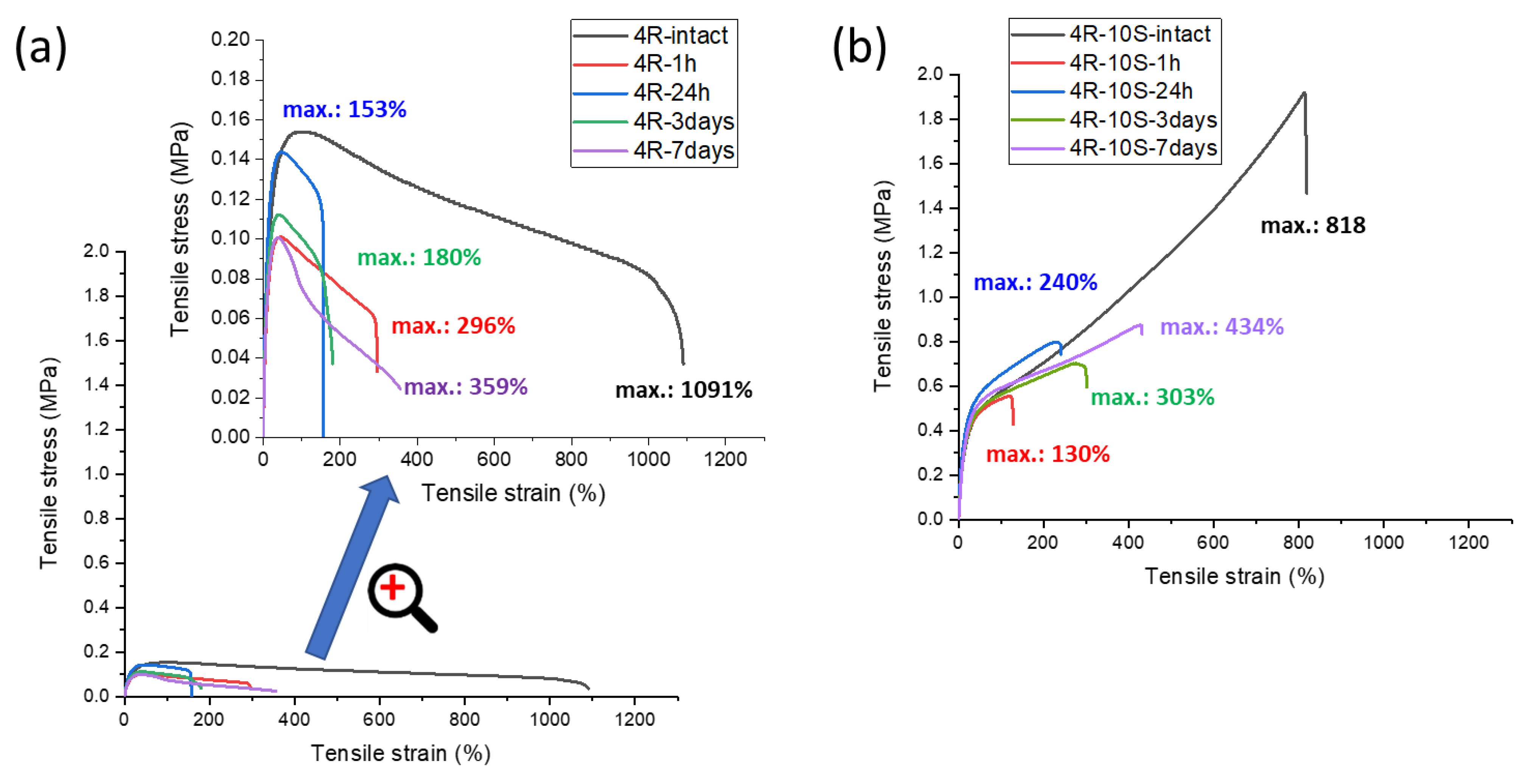
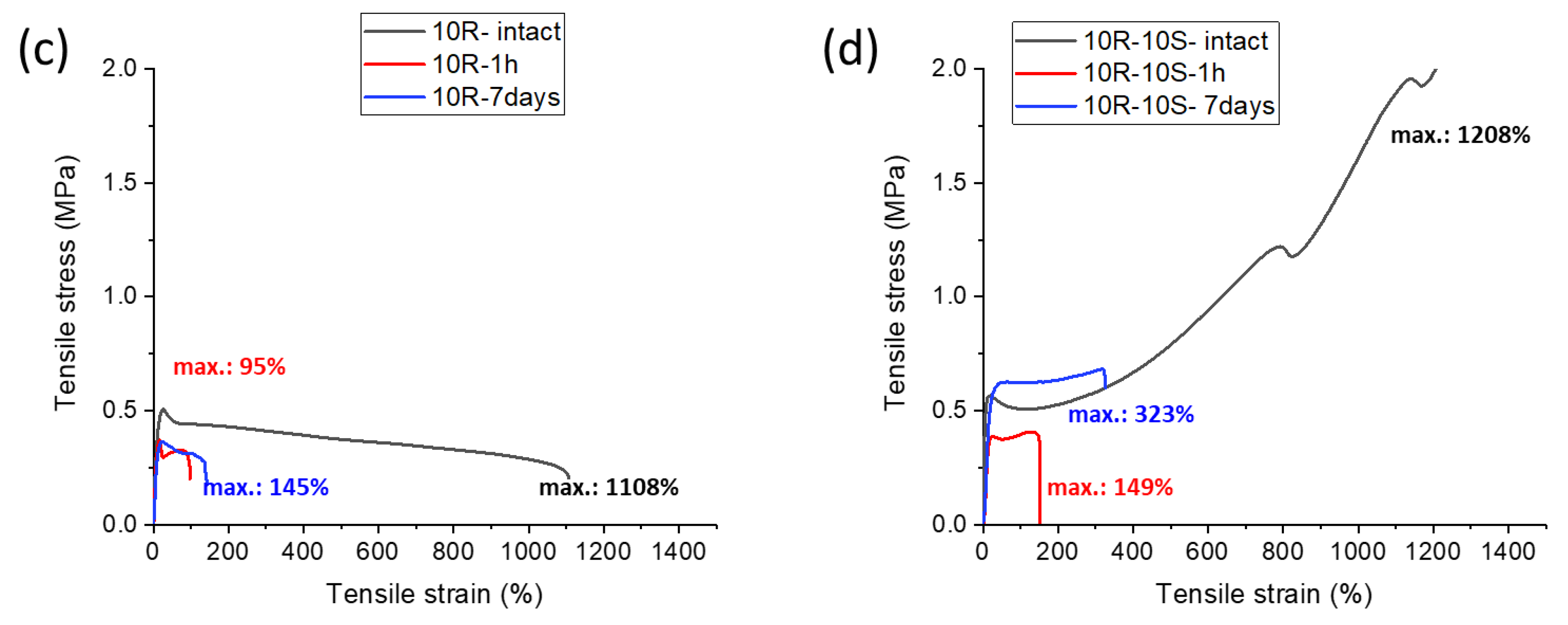
2.5. Self-Healing of Disrupted Samples and Its Efficiency
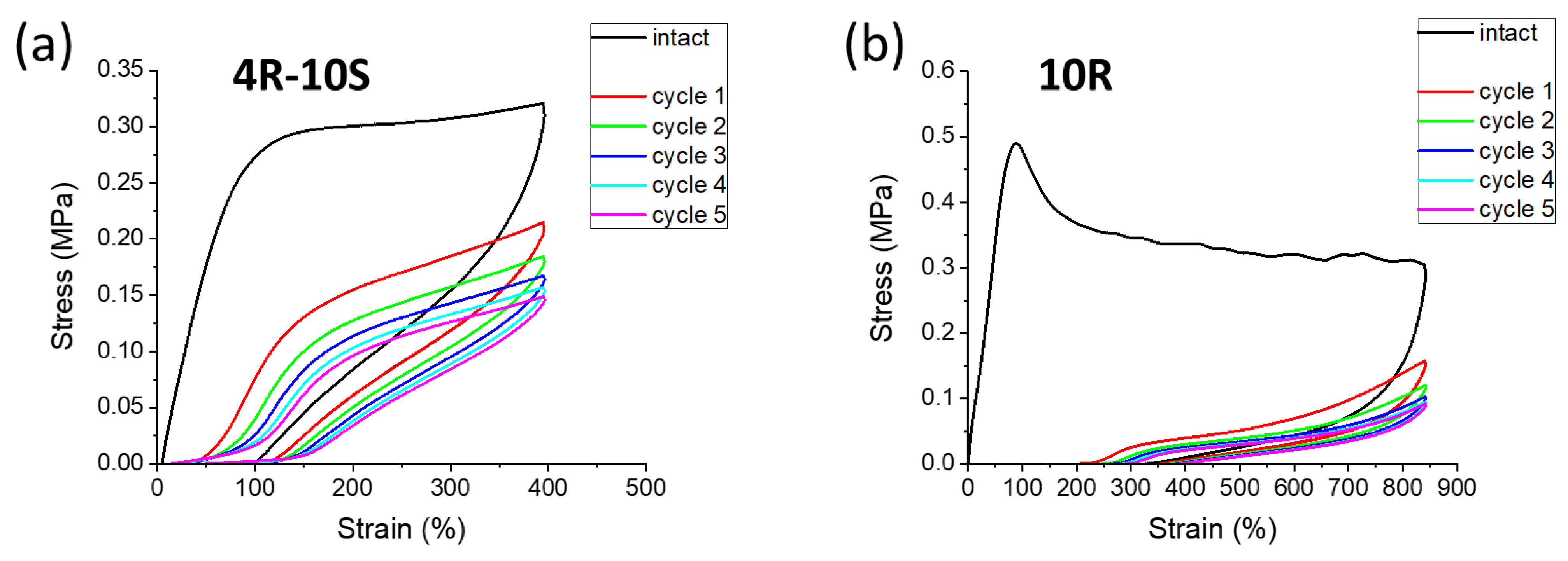
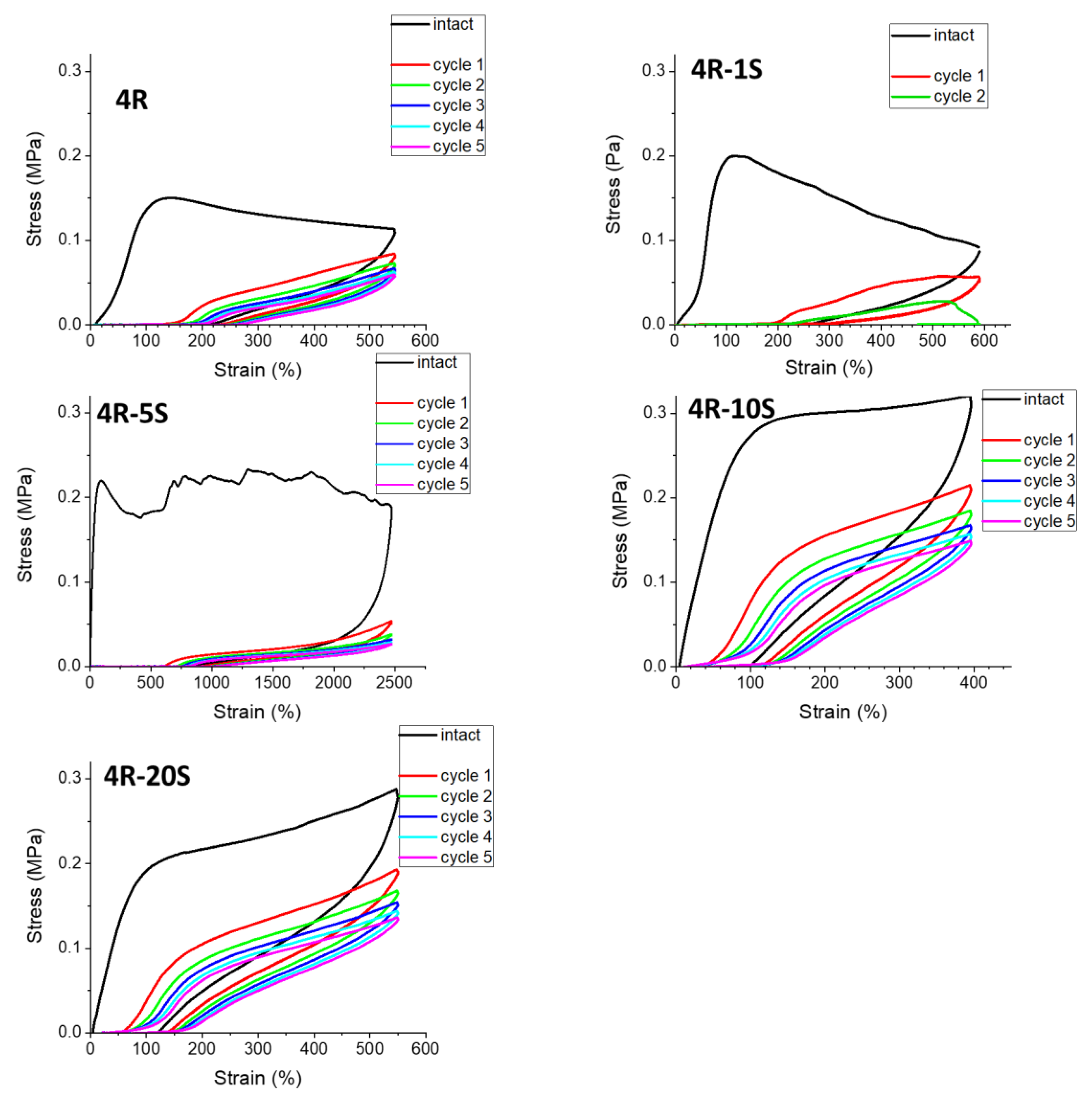
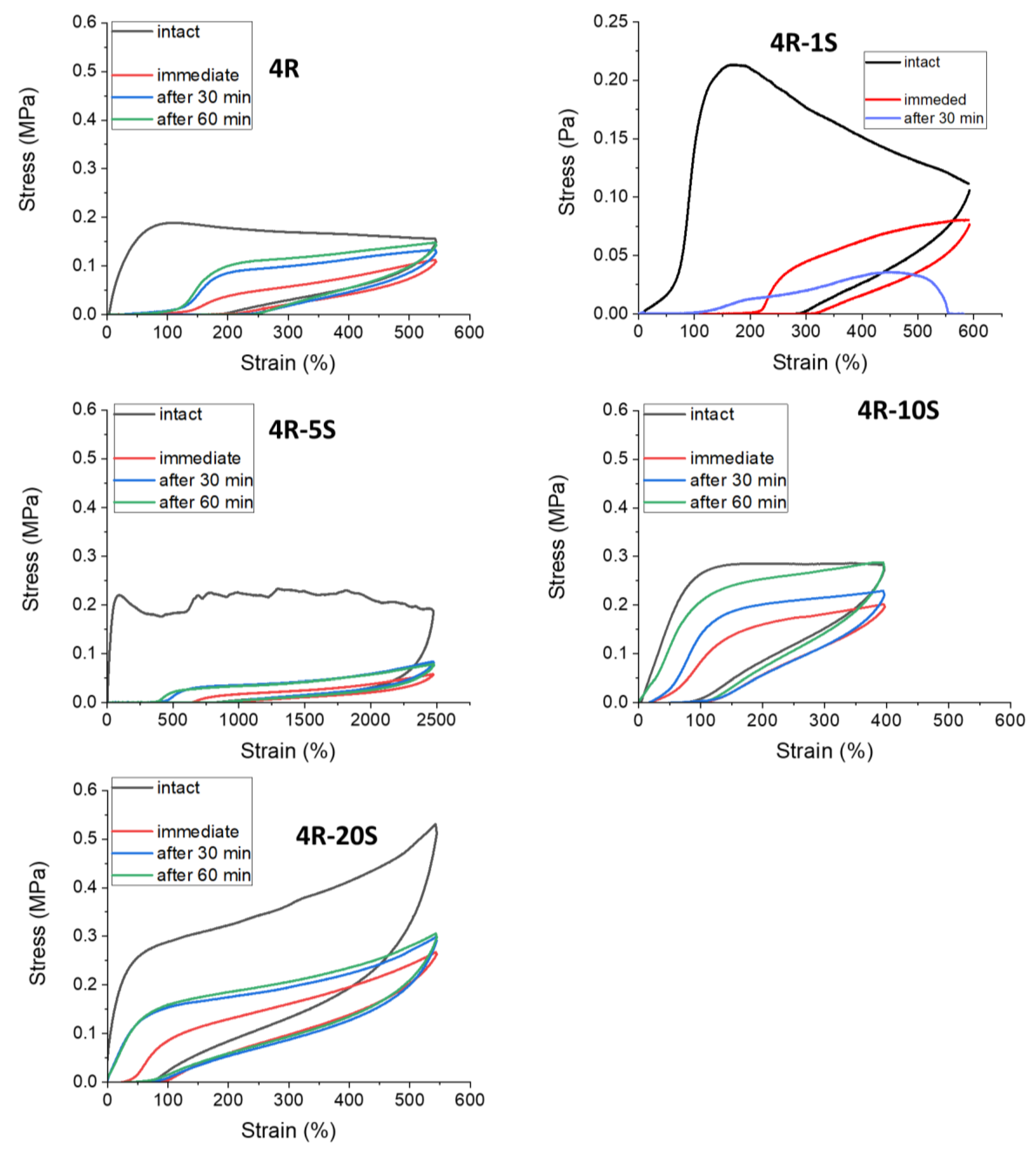
2.6. Elasticity vs. Creep after Large Stretching Deformation: Cyclic Loading Tests
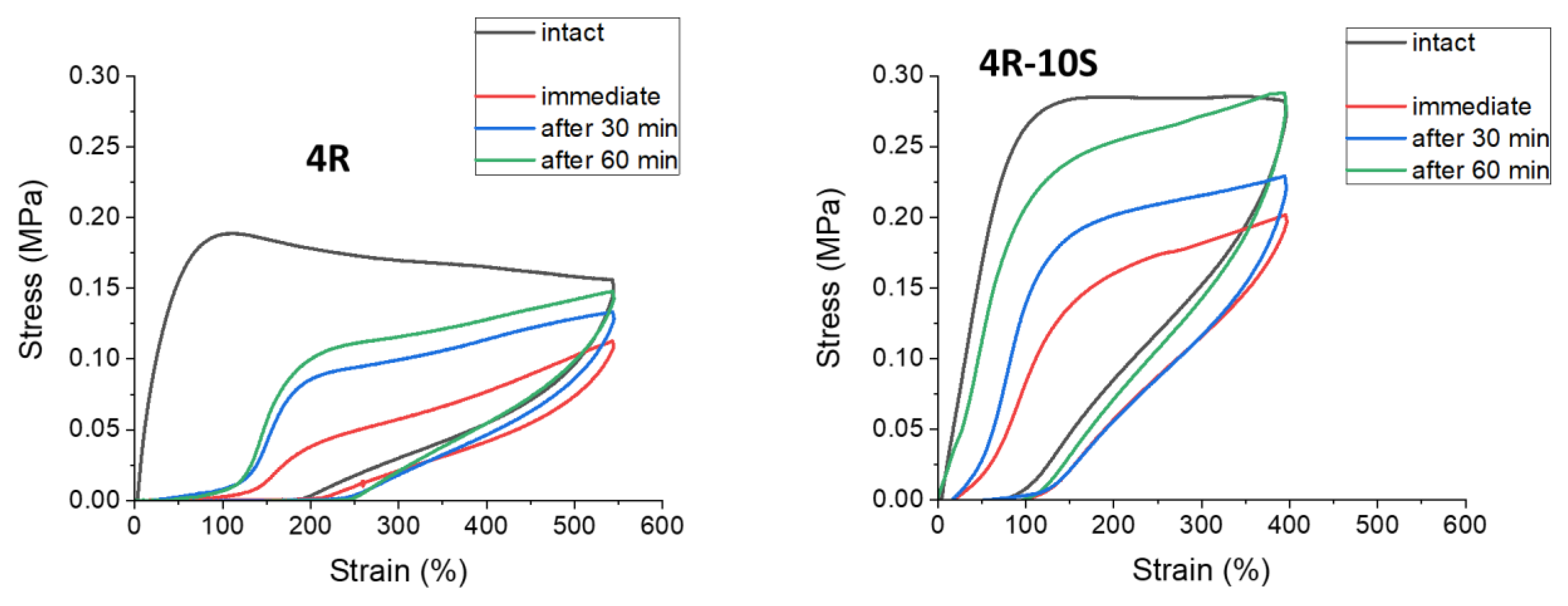
2.7. Internal Self-Healing: Differently Fast Self-Recovery of Mechanical Properties after Large Stretching Deformation
3. Conclusions
- -
- Novel solvent-free nanocomposite super-elastomers based on the copolymer matrix poly(methoxyethyl acrylate-co-sodium methacrylate) physically crosslinked by clay nano-platelets (‘poly[MEA-co-SMA]/clay’) were synthesized;
- -
- Depending on the SMA content, the super-elastomers were predominantly hydrophobic, water-swelling but stable against dissolution, or fully water-soluble and hence solution-processible;
- -
- Generally, in the dry state the SMA co-monomer introduces a tremendous increase in tensile strength, combined with an increase in toughness, while ultra-extensibility similar to the simpler ‘poly[MEA]/clay’ nanocomposites is preserved;
- -
- Variation of composition parameters makes possible to obtain a very wide range of product properties, including extreme ultra-extensibility, or high stiffness in rubbery state combined with more moderate super-extensibility, or very different values of tensile strength;
- -
- The SMA co-monomer introduces a great improvement in self-healing ability of specimens which were cut, in comparison to the simpler poly[MEA]/clay systems; the best results were achieved with 10 mol% of SMA in the matrix and 4 wt.% of nano-clay filler (macro-crosslinker); the regeneration was 53% of the original tensile curve after 1 week at simple conditions, in case of the mentioned most attractive product; variation of SMA content switches the elastic behavior at large deformations between plasto-elastic, elasto-plastic, and viscous elastic type;
- -
- Self-recovery of mechanical damage after large deformations was tremendously improved by the SMA co-monomer, and even complete self-recovery was achieved; the best results were obtained with 10 mol% of SMA and not-too-low clay content;
- -
- Elucidation of structure–property relationships explained the principle of the tremendous effect of the SMA co-monomer on mechanical and self-healing properties: it is based on the formation of multiplets (nano-aggregates) of the ionic SMA units, which are phase-separated in the hydrophobic polyMEA matrix: at low SMA contents, the multiplets can form strong, highly isolated, and irreversibly dissociating physical crosslinks which support more plasticity; or, at higher SMA contents, the multiplets can dynamically exchange SMA units, because they are less separated, which in turn supports rapid self-healing;
- -
- The studied super-elastomers are attractive for potential applications as advanced self-healing materials for engineering, robotics, medical or implant technology.
4. Materials and Methods
4.1. Materials
4.2. Synthesis of the Nanocomposite Elastomers
4.3. Self-Healing Tests
4.4. Characterization
4.4.1. NMR
4.4.2. TEM
4.4.3. Thermo-Mechanical Properties (DMTA)
4.4.4. DSC Analyses
4.4.5. Simple Tensile Tests
4.4.6. Cyclic Tensile Tests
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ulrich, H. Introduction to Industrial Polymers; Macmillan Publishing Company Inc.: New York, NY, USA, 1982; ISBN10: 0029497906, ISBN13: 9780029497906. [Google Scholar]
- Haraguchi, K.; Takehisa, T. Nanocomposite Hydrogels: A Unique Organic–Inorganic Network Structure with Extraordinary Mechanical, Optical, and Swelling/De-swelling Properties. Adv. Mater. 2002, 14, 1120–1124. [Google Scholar] [CrossRef]
- Haraguchi, K. Synthesis and properties of soft nanocomposite materials with novel organic/inorganic network structures. Polym. J. 2011, 43, 223–241. [Google Scholar] [CrossRef]
- Xia, L.W.; Xie, R.; Ju, X.J.; Wang, W.; Chen, Q.; Chu, L.Y. Nano-structured smart hydrogels with rapid response and high elasticity. Nat. Commun. 2013, 4, 2226. [Google Scholar] [CrossRef]
- Goswami, S.K.; McAdam, C.J.; Hanton, L.R.; Moratti, S.C. Hyperelastic Tough Gels through Macrocross-Linking. Macromol. Rapid Commun. 2017, 38, 1700103. [Google Scholar] [CrossRef]
- Sun, G.; Li, Z.; Liang, R.; Weng, L.T.; Zhang, L. Super stretchable hydrogel achieved by non-aggregated spherulites with diameters < 5nm. Nat. Commun. 2016, 7, 12095. [Google Scholar] [CrossRef]
- Gaharwar, A.K.; Dammu, S.A.; Canter, J.M.; Wu, C.J.; Schmidt, G. Highly Extensible, Tough, and Elastomeric Nanocomposite Hydrogels from Poly(ethylene glycol) and Hydroxyapatite Nanoparticles. Biomacromolecules 2011, 12, 1641–1650. [Google Scholar] [CrossRef]
- Wang, Q.; Li, L.; Li, Z.; Guo, S.; Sun, G. Environmentally Stable Polymer Gels with Super Deformability and High Recoverability Enhanced by Sub-5 nm Particles in the Nonvolatile Solvent. J. Polym. Sci. Part B: Polym. Phys. 2019, 57, 713–721. [Google Scholar] [CrossRef]
- Liu, R.; Liang, S.; Tang, X.Z.; Yan, D.; Li, X.; Yu, Z.Z. Tough and highly stretchable graphene oxide/polyacrylamide nanocomposite hydrogels. J. Mater. Chem. 2012, 22, 14160–14167. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, G. Novel Nanocomposite Hydrogels Consisting of Layered Double Hydroxide with Ultrahigh Tensibility and Hierarchical Porous Structure at Low Inorganic Content. Adv. Mater. 2014, 26, 5950–5956. [Google Scholar] [CrossRef]
- Haraguchi, K.; Uyama, K.; Tanimoto, H. Self-healing in Nanocomposite Hydrogels. Macromol. Rapid Commun. 2011, 32, 1253–1258. [Google Scholar] [CrossRef]
- Gao, G.; Du, G.; Sun, Y.; Fu, J. Self-Healable, Tough, and Ultrastretchable Nanocomposite Hydrogels Based on Reversible Polyacrylamide/Montmorillonite Adsorption. ACS Appl. Mater. Interfaces 2015, 7, 5029–5037. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, K.; Li, H.J.; Masuda, K.; Takehisa, T.; Elliott, E. Mechanism of Forming Organic/Inorganic Network Structures during In-situ Free-Radical Polymerization in PNIPA−Clay Nanocomposite Hydrogels. Macromolecules 2005, 38, 3482–3490. [Google Scholar] [CrossRef]
- Haraguchi, K.; Farnworth, R.; Ohbayashi, A.; Takehisa, T. Compositional effects on mechanical properties of nanocomposite hydrogels composed of poly (N,N-dimethylacrylamide) and clay. Macromolecules 2003, 36, 5732–5741. [Google Scholar] [CrossRef]
- Haraguchi, K.; Li, H.J. Mechanical properties and structure of polymer–Clay nanocomposite gels with high clay content. Macromolecules 2006, 39, 1898–1905. [Google Scholar] [CrossRef]
- Yu, J.C.; Tonpheng, B.; Grobner, G.; Andersson, O. A MWCNT/Polyisoprene Composite Reinforced by an Effective Load Transfer Reflected in the Extent of Polymer Coating. Macromolecules 2012, 45, 2841–2849. [Google Scholar] [CrossRef]
- Hakimelahi, H.R.; Hu, L.; Rupp, B.B.; Coleman, M.R. Synthesis and characterization of transparent alumina reinforced polycarbonate nanocomposite. Polymer 2010, 51, 2494–2502. [Google Scholar] [CrossRef]
- Zhou, W.; Yu, Y.; Chen, H.; DiSalvo, F.J.; Abruna, H.D. Yolk–Shell Structure of Polyaniline-Coated Sulfur for Lithium–Sulfur Batteries. J. Am. Chem. Soc. 2013, 135, 16736–16743. [Google Scholar] [CrossRef]
- Matteucci, S.; van Wagner, E.; Freeman, B.D.; Swinnea, S.; Sakaguchi, T.; Masuda, T. Desilylation of Substituted Polyacetylenes by Nanoparticles. Macromolecules 2007, 40, 3337–3347. [Google Scholar] [CrossRef]
- Strachota, A.; Ribot, F.; Matějka, L.; Whelan, P.; Starovoytova, L.; Plestil, J.; Steinhart, M.; Slouf, M.; Hromadkova, J.; Kovarova, J.; et al. Preparation of novel, nanocomposite stannoxane-based organic-inorganic epoxy polymers containing ionic bonds. Macromolecules 2012, 45, 221–237. [Google Scholar] [CrossRef]
- Strachota, A.; Rodzen, K.; Ribot, F.; Perchacz, M.; Trchová, M.; Steinhart, M.; Starovoytova, L.; Slouf, M.; Strachota, B. Tin-based “super-POSS” building blocks in epoxy nanocomposites with highly improved oxidation resistance. Polymer 2014, 55, 3498–3515. [Google Scholar] [CrossRef]
- Strachota, A.; Rodzeń, K.; Ribot, F.; Trchová, M.; Steinhart, M.; Starovoytova, L.; Pavlova, E. Behavior of Tin-Based “Super-POSS” Incorporated in Different Bonding Situations in Hybrid Epoxy Resins. Macromolecules 2014, 47, 4266–4287. [Google Scholar] [CrossRef]
- Rodzeń, K.; Strachota, A.; Ribot, F.; Šlouf, M. Effect of network mesh size on the thermo-mechanical properties of epoxy nanocomposites with the heavier homologue of POSS, the inorganic butylstannoxane cages. Eur. Polym. J. 2014, 57, 169–181. [Google Scholar] [CrossRef]
- Strachota, A.; Rodzeń, K.; Raus, V.; Ribot, F.; Janata, M.; Pavlova, E. Incorporation and chemical effect of Sn-POSS cages in poly(ethyl methacrylate). Eur. Polym. J. 2015, 68, 366–378. [Google Scholar] [CrossRef]
- Rodzeń, K.; Strachota, A.; Ribot, F.; Matějka, L.; Kovářová, J.; Trchová, M.; Šlouf, M. Reactivity of the tin homolog of POSS, butylstannoxane dodecamer, in oxygen-induced crosslinking reactions with an organic polymer matrix: Study of long-time behavior. Polym. Degrad. Stab. 2015, 118, 147–166. [Google Scholar] [CrossRef]
- Rodzeń, K.; Strachota, A.; Raus, V.; Pavlova, E. Polyhedral oligomeric butyl stannoxane cages (Sn-POSS) as oxidation activated linear repairing units or crosslinking nano-building blocks, depending on structure of the polymer matrix. Polym. Degrad. Stab. 2017, 142, 1–20. [Google Scholar] [CrossRef]
- Strachota, B.; Strachota, A.; Horodecka, S.; Steinhart, M.; Kovářová, J.; Pavlova, E.; Ribot, F. Polyurethane nanocomposites containing the chemically active inorganic Sn-POSS cages Reactive and Functional Polymers. React. Funct. Polym. 2019, 143, 104338. [Google Scholar] [CrossRef]
- Rao, Y.Q.; Chen, S. Molecular Composites Comprising TiO2 and Their Optical Properties. Macromolecules 2008, 41, 4838–4844. [Google Scholar] [CrossRef]
- Miniewicz, A.; Girones, J.; Karpinski, P.; Mossety-Leszczak, B.; Galina, H.; Dutkiewicz, M. Photochromic and nonlinear optical properties of azo-functionalized POSS nanoparticles dispersed in nematic liquid crystals. J. Mater. Chem. C 2014, 2, 432–440. [Google Scholar] [CrossRef]
- Kim, H.; Abdala, A.A.; Macosko, C.W. Graphene/Polymer Nanocomposites. Macromolecules 2010, 43, 6515–6530. [Google Scholar] [CrossRef]
- Depa, K.; Strachota, A.; Šlouf, M.; Brus, J.; Cimrová, V. Synthesis of conductive doubly filled poly(N-isopropylacrylamide)-polyaniline-SiO2 hydrogels. Sens. Actuators B-Chem. 2017, 244, 616–634. [Google Scholar] [CrossRef]
- Robbes, A.S.; Jestin, J.; Meneau, F.; Dalmas, F.; Sandre, O.; Perez, J.; Boue, F.; Cousin, F. Homogeneous Dispersion of Magnetic Nanoparticles Aggregates in a PS Nanocomposite: Highly Reproducible Hierarchical Structure Tuned by the Nanoparticles’ Size. Macromolecules 2010, 43, 5785–5796. [Google Scholar] [CrossRef]
- Mossety-Leszczak, B.; Strachota, B.; Strachota, A.; Steinhart, M.; Šlouf, M. The orientation-enhancing effect of diphenyl aluminium phosphate nanorods in a liquid-crystalline epoxy matrix ordered by magnetic field. Eur. Polym. J. 2015, 72, 238–255. [Google Scholar] [CrossRef]
- Maji, P.K.; Das, N.K.; Bhowmick, A.K. Preparation and properties of polyurethane nanocomposites of novel architecture as advanced barrier materials. Polymer 2010, 51, 1100–1110. [Google Scholar] [CrossRef]
- Spirkova, M.; Brus, J.; Brozova, L.; Strachota, A.; Baldrian, J.; Urbanova, M.; Kotek, J.; Strachotova, B.; Slouf, M. A view from inside onto the surface of self-assembled nanocomposite coatings. Prog. Org. Coat. 2008, 61, 145–155. [Google Scholar] [CrossRef]
- Yan, N.; Buonocore, G.; Lavorgna, M.; Kaciulis, S.; Balijepalli, S.K.; Zhan, Y.H.; Xia, H.S.; Ambrosio, L. The role of reduced graphene oxide on chemical, mechanical and barrier properties of natural rubber composites. Compos. Sci. Technol. 2014, 102, 74–81. [Google Scholar] [CrossRef]
- Daniel, W.F.M.; Burdynska, J.; Vatankhah-Varnoosfaderani, M.; Matyjaszewski, K.; Paturej, J.; Rubinstein, M.; Dobrynin, A.V.; Sheiko, S.S. Solvent-free, supersoft and superelastic bottlebrush melts and networks. Nat. Mater. 2016, 15, 183–190. [Google Scholar] [CrossRef]
- Haraguchi, K.; Ebato, M.; Takehisa, T. Polymer–Clay Nanocomposites Exhibiting Abnormal Necking Phenomena Accompanied by Extremely Large Reversible Elongations and Excellent Transparency. Adv. Mater. 2006, 18, 2250–2254. [Google Scholar] [CrossRef]
- Haraguchi, K.; Masatoshi, S.; Kotobuki, N.; Murata, K. Thermoresponsible Cell Adhesion/Detachment on Transparent Nanocomposite Films Consisting of Poly(2-Methoxyethyl Acrylate) and Clay. J. Biomater. Sci. 2011, 22, 2389–2406. [Google Scholar] [CrossRef]
- Haraguchi, K. Development of Soft Nanocomposite Materials and Their Applications in Cell Culture and Tissue Engineering. J. Stem. Cells Regen. Med. 2012, 8, 2–11. Available online: https://www.pubstemcell.com/monthly/008010200002.htm (accessed on 15 September 2022).
- Tanaka, M.; Motomura, T.; Kawada, M.; Anzai, T.; Kasori, Y.; Onishi, M.; Shiroya, T.; Shimura, K.; Mochizuki, A. Blood compatible aspects of poly(2-methoxyethylacrylate) (PMEA)—relationship between protein adsorption and platelet adhesion on PMEA surface. Biomaterials 2000, 21, 1471–1481. [Google Scholar] [CrossRef]
- Hayashi, M.; Noro, A.; Matsushita, Y. Highly Extensible Supramolecular Elastomers with Large Stress Generation Capability Originating from Multiple Hydrogen Bonds on the Long Soft Network Strands. Macromol. Rapid Commun. 2016, 37, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Lutecki, M.; Strachotová, B.; Uchman, M.; Brus, J.; Pleštil, J.; Šlouf, M.; Strachota, A.; Matějka, L. Thermosensitive PNIPA-based organic-inorganic hydrogels. Polym. J. 2006, 38, 527–541. [Google Scholar] [CrossRef]
- Strachotová, B.; Strachota, A.; Uchman, M.; Šlouf, M.; Brus, J.; Pleštil, J.; Matějka, L. Super porous organic–inorganic poly(N-isopropylacrylamide)-based hydrogel with a very fast temperature response. Polymer 2007, 48, 1471–1482. [Google Scholar] [CrossRef]
- Strachota, B.; Šlouf, M.; Matějka, L. Tremendous reinforcing, pore-stabilizing and response-accelerating effect of in situ generated nanosilica in thermoresponsive poly(N-isopropylacrylamide) cryogels. Polym. Int. 2017, 66, 1510–1521. [Google Scholar] [CrossRef]
- Depa, K.; Strachota, A.; Šlouf, M.; Brus, J. Poly(N-isopropylacrylamide)-SiO2 nanocomposites interpenetrated by starch: Stimuli-responsive hydrogels with attractive tensile properties. Eur. Polym. J. 2017, 88, 349–372. [Google Scholar] [CrossRef]
- Huerta-Angeles, G.; Hishchak, K.; Strachota, A.; Strachota, B.; Šlouf, M.; Matějka, L. Super-porous nanocomposite PNIPAm hydrogels reinforced with titania nanoparticles, displaying a very fast temperature response as well as pH-sensitivity. Eur. Polym. J. 2014, 59, 341–352. [Google Scholar] [CrossRef]
- Strachota, B.; Matejka, L.; Zhigunov, A.; Konefal, R.; Spevacek, J.; Dybal, J.; Puffr, R. Poly(N-isopropylacrylamide)-clay based hydrogels controlled by the initiating conditions: Evolution of structure and gel formation. Soft Matter 2015, 11, 9291–9306. [Google Scholar] [CrossRef]
- Strachota, B.; Hodan, J.; Matějka, L. Poly(N-isopropylacrylamide)-clay hydrogels: Control of mechanical properties and structure by the initiating conditions of polymerization. Eur. Polym. J. 2016, 77, 1–15. [Google Scholar] [CrossRef]
- Strachota, B.; Matějka, L.; Sikora, A.; Spěváček, J.; Konefal, R.; Zhigunov, A.; Šlouf, M. Insight into the cryopolymerization to form a poly(N-isopropylacrylamide)/clay macroporous gel: Structure and phase evolution. Soft Matter 2017, 13, 1244–1256. [Google Scholar] [CrossRef]
- Strachota, B.; Šlouf, M.; Hodan, J.; Matějka, L. Advanced two-step cryopolymerization to form superporous thermosensitive PNIPA/clay gels with unique mechanical properties and ultrafast swelling-deswelling kinetics. Colloid Polym. Sci. 2018, 296, 753–769. [Google Scholar] [CrossRef]
- Strachota, B.; Strachota, A.; Steinhart, M.; Šlouf, M.; Hodan, J. Ultra-extensible solvent-free elastomers based on nanocomposite poly(2-methoxyethylacrylate)/clay xerogels. J. Appl. Polym. Sci. 2021, 138, e49836. [Google Scholar] [CrossRef]
- Byś, K.; Strachota, B.; Strachota, A.; Pavlova, E.; Steinhart, M.; Mossety-Leszczak, B.; Zając, W. Novel Tough and Transparent Ultra-Extensible Nanocomposite Elastomers Based on Poly(2-methoxyethylacrylate) and Their Switching between Plasto-Elasticity and Viscoelasticity. Polymers 2021, 13, 4254. [Google Scholar] [CrossRef] [PubMed]
- Strachota, B.; Strachota, A.; Gąsior, G.; Šlouf, M. High-strength nanocomposite self-regenerating hydrogels reinforced by additional crosslinking with trivalent metal cations. J. Polym. Res. 2021, 28, 211. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | H2O | Clay RDS | MEA | n MEA | SMA | n SMA | TEMED | n TEMED | 1% APS aq | n APS |
|---|---|---|---|---|---|---|---|---|---|---|
| g | g | g | mmol | g | mmol | g | mmol | g | mmol | |
| PMEA-matrix | 42.312 | 0 | 5 | 38.42 | 0 | 0 | 0.0625 | 0.538 | 3.814 | 0.167 |
| 0R-10S * | 47.108 | 0 | 5 | 38.42 | 0.461 | 4.269 | 0.0695 | 0.597 | 4.238 | 0.186 |
| 2R | 42.301 | 0.111 | 5 | 38.42 | 0 | 0 | 0.0625 | 0.538 | 3.814 | 0.167 |
| 4R | 42.290 | 0.229 | 5 | 38.42 | 0 | 0 | 0.0625 | 0.538 | 3.814 | 0.167 |
| 10R | 42.251 | 0.614 | 5 | 38.42 | 0 | 0 | 0.0625 | 0.538 | 3.814 | 0.167 |
| 2R-10S * | 47.096 | 0.121 | 5 | 38.42 | 0.461 | 4.269 | 0.0695 | 0.598 | 4.238 | 0.186 |
| 4R-1S | 42.723 | 0.236 | 5 | 38.42 | 0.042 | 0.388 | 0.0631 | 0.543 | 3.852 | 0.168 |
| 4R-5S | 44.560 | 0.239 | 5 | 38.42 | 0.219 | 2.022 | 0.0658 | 0.566 | 4.015 | 0.176 |
| 4R-10S * | 47.083 | 0.250 | 5 | 38.42 | 0.461 | 4.269 | 0.0695 | 0.598 | 4.238 | 0.186 |
| 4R-20S * | 53.075 | 0.276 | 5 | 38.42 | 1.038 | 9.605 | 0.0781 | 0.672 | 4.767 | 0.209 |
| 10R-10S | 47.041 | 0.671 | 5 | 38.42 | 0.461 | 4.269 | 0.0695 | 0.598 | 4.238 | 0.186 |
| Sample | Tg (1) [°C] | Change in Heat Capacity at Tg (1) [J g−1 K−1] | Tg (2) [°C] | Change in Heat Capacity at Tg (2) [J g−1 K−1] |
|---|---|---|---|---|
| neat polyMEA | (−34.75) −30.75 | (0.602) 0.604 | - | - |
| 2R | (−35.88) −32.33 | (0.638) 0.673 | - | - |
| 4R | (−34.98) −31.18 | (0.620) 0.617 | - | - |
| 10R | (−34.70) −31.26 | (0.465) 0.465 | - | - |
| 2R-10S | (−33.23) −31.88 | (0.283) 0.267 | (−11.94) +1.28 (flat) | (0.331) 0.242 |
| 4R-10S | (−34.80) −32.68 | (0.284) 0.244 | (−21.01) +0.48 (flat) | (0.335) 0.221 |
| 10R-10S | (−21.31) −15.58 | (0.548) 0.410 | (no second step, but broadening and shift in second scan) | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strachota, B.; Strachota, A.; Byś, K.; Pavlova, E.; Hodan, J.; Mossety-Leszczak, B. Self-Healing and Super-Elastomeric PolyMEA-co-SMA Nanocomposites Crosslinked by Clay Platelets. Gels 2022, 8, 657. https://doi.org/10.3390/gels8100657
Strachota B, Strachota A, Byś K, Pavlova E, Hodan J, Mossety-Leszczak B. Self-Healing and Super-Elastomeric PolyMEA-co-SMA Nanocomposites Crosslinked by Clay Platelets. Gels. 2022; 8(10):657. https://doi.org/10.3390/gels8100657
Chicago/Turabian StyleStrachota, Beata, Adam Strachota, Katarzyna Byś, Ewa Pavlova, Jiří Hodan, and Beata Mossety-Leszczak. 2022. "Self-Healing and Super-Elastomeric PolyMEA-co-SMA Nanocomposites Crosslinked by Clay Platelets" Gels 8, no. 10: 657. https://doi.org/10.3390/gels8100657
APA StyleStrachota, B., Strachota, A., Byś, K., Pavlova, E., Hodan, J., & Mossety-Leszczak, B. (2022). Self-Healing and Super-Elastomeric PolyMEA-co-SMA Nanocomposites Crosslinked by Clay Platelets. Gels, 8(10), 657. https://doi.org/10.3390/gels8100657