1. Introduction
Vesicles based on lipids are recognized as promising non-viral vectors for drug or gene delivery purposes [
1,
2,
3,
4,
5]. In particular, phospholipid liposomes are considered as suitable vehicles for pharmaceutical compounds due to their biodegradable and biocompatible nature, which is combined with low toxicity levels [
6]. In such liposome/drug formulations, the nature of the interactions between the guest molecules and the lipid vesicle is of paramount significance since it is intimately related to the ability of the carrier to minimize the loss of the bioactive cargo during the transfer to the desired destination, as well as to the mechanism of its liposomal escape at the target site [
7,
8].
Molecular simulations have started playing an increasingly important role in the description of the interactions of bioactive molecules with lipid layers [
9,
10,
11,
12], due to their ability to provide information at the molecular or even at the atomic level. Among other model phospholipid layers, dipalmitoylphosphatidylglycerol (DPPG)-based systems have been recently studied in drug/liposome formulations [
13]. DPPG membranes (formed by a double layer of lipids, i.e., a bilipid layer) carry a negative charge per lipid at physiological pH conditions [
14,
15] which renders them good candidates when complexation with cationic bioactive compounds is desired [
13,
14,
16]. This attribute of DPPG lipids can be exploited in more complex drug/liposome formulations, where optimization of the drug loading conditions is promoted by the presence of multifunctional molecules such as hyperbranched polymers [
4,
17,
18].
In this work we examine by means of fully atomistic molecular dynamics simulations, the interactions of a complex comprised by a functionalized (PEGylated) hyperbranched polymer (HBP) and an anticancer drug, doxorubicin, with a model bilipid DPPG membrane in an aqueous environment. Structural and dynamic characteristics of the hyperbranched-polymer/doxorubicin aqueous solution have been investigated in our previous work [
19]. We henceforth aim at elucidating the effects of the presence of the DPPG bilayer to the drug spatial distribution with respect to the polymer and the lipid membrane, the nature of the drug/DPPG interactions, the effective charge distributions related to possible overcharging phenomena of the polymer and of the bilipid DPPG layer, and the changes in the translational behavior of the drug molecules imparted by the presence of the bilipid membrane.
2. Materials and Methods
Initial configuration of the DPPG bilipid membrane was taken from a pre-equilibrated system of a several hundred ns long simulation, from a publicly available repository [
20]. Pre-equilibration of the hydrated DPPG membranes were performed by Jämbeck and collaborators [
21,
22,
23] in explicit TIP3P [
24] water solution.
Figure 1 illustrates a single DPPG lipid, a doxorubicin molecule, and the structure of the PEGylated hyperbranched polymer.
Figure 2 depicts the equilibrated DPPG membrane comprised by 128 lipids. As a next step, the bilipid membrane was combined with the HBP/Doxorubicin complex that was taken from our previous study [
19] with the aid of the VEGA ZZ software [
25]. The HBP/Doxorubicin complex was initially placed on top of one side of the DPPG bilayer with a separation of approximately 65 Å between the center of mass of the HBP and that of the membrane (no constraints have been imposed to any of the constituents during the equilibration or the production runs). For comparison purposes, we have kept exactly the same number of neutral and protonated drug molecules and the same ionic strength of 165 mM as in Reference [
19].
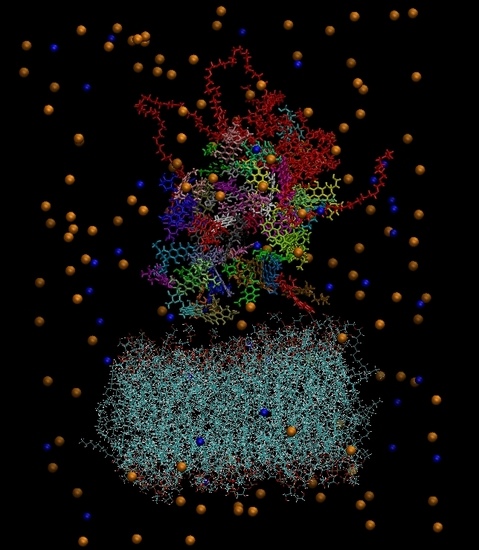
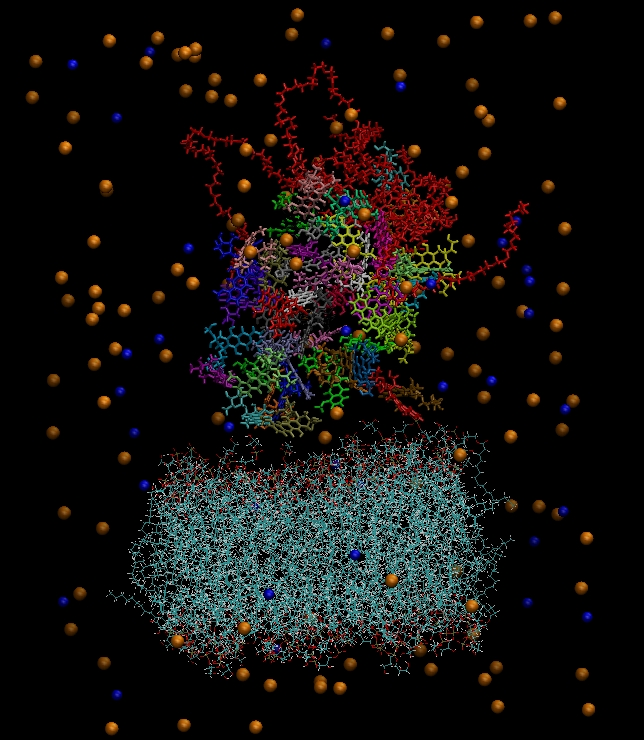
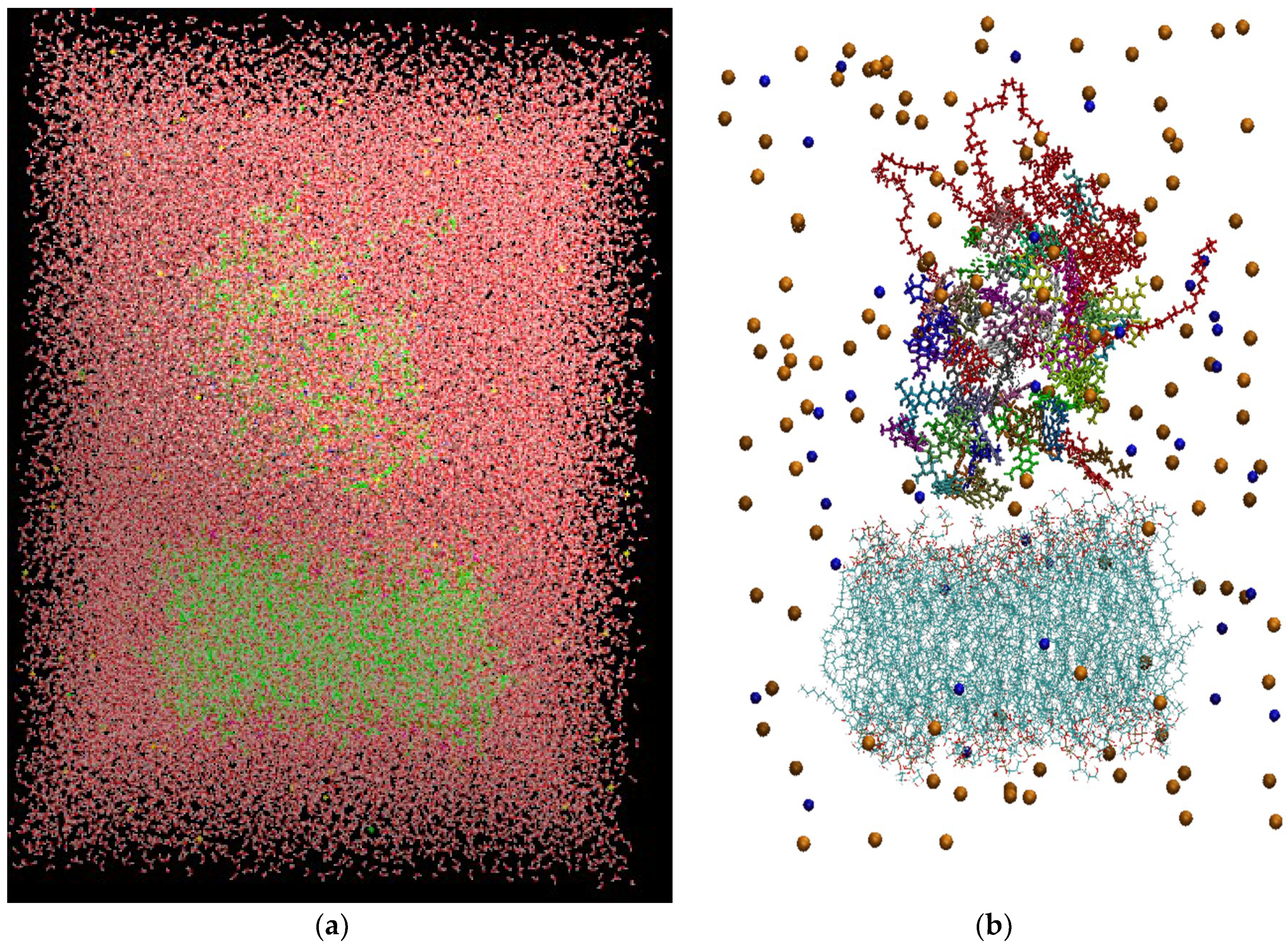
Figure 3 depicts the initial configuration of the bilipid/hyperbranched/drugs/counterions system, with (
a) and without (
b) the water molecules. The number of the different components comprising the examined system are presented in
Table 1.
The forcefield parameters for the hyperbranched polymer and the drug molecules were taken from the generalized Amber forcefield (GAFF) [
26] and for the water molecules from the TIP3P model [
24] as described in Reference [
19]. For the bilipid membrane, the GAFF lipid set of parameters was utilized, which was developed specifically for the description of lipids [
27]. All simulations were performed by the aid of the NAMD simulation package [
28] with a timestep of 1 fs and a saving frequency of 2 ps, using periodic boundary conditions. Temperature control was performed using the Langevin method (with a damping coefficient of 5 ps
−1) [
29]. Electrostatic interactions were calculated via the particle mesh Ewald (PME) scheme [
30], while the distance cutoff for the van der Waals interactions was set at 12 Å. The percentage of the protonated doxorubicin molecules was calculated through the Henderson−Hasselbalch relation [
31] taking into account the pK
a of the drug’s ionizable group at 37 °C and considering physiological pH conditions. An appropriate amount of chlorine and sodium ions was added for the overall neutralization of the system and for accomplishing an ionic strength of 165 mM [
32].
More detailed information regarding the construction and the equilibration of the hyperbranched polymer/drug complex can be found in Reference [
19]. Following the construction of the system, energy minimization with steepest descent and conjugate gradient methods was applied, prior to 40 ns long MD equilibration runs in the canonical (NVT) ensemble at T = 310 K. This procedure ensured stabilization of energetic (total energy), thermodynamic (pressure), and conformational properties (i.e., average size of the PEGylated hyperbranched polymer) of the polymeric component of the system. The equilibrium area per lipid was approximately 75 Å
2 and the parallel orientation of the lipids was found to persist throughout the simulation. Production runs were also performed in the NVT ensemble and at the same (T = 310 K) temperature.
3. Results
The analysis performed in the simulation data aimed at investigating the spatial arrangement of the drug molecules with respect to the hyperbranched carrier and the negatively charged DPPG membrane, the degree of their association with the latter, and the driving forces responsible for the observed association.
3.1. Spatial Distribution of Drug Molecules
To examine the spatial arrangement of the drug molecules with respect to the hyperbranched nanoparticle and the bilipid membrane, we have calculated relevant average distances between centers of mass as well as relevant mass distributions.
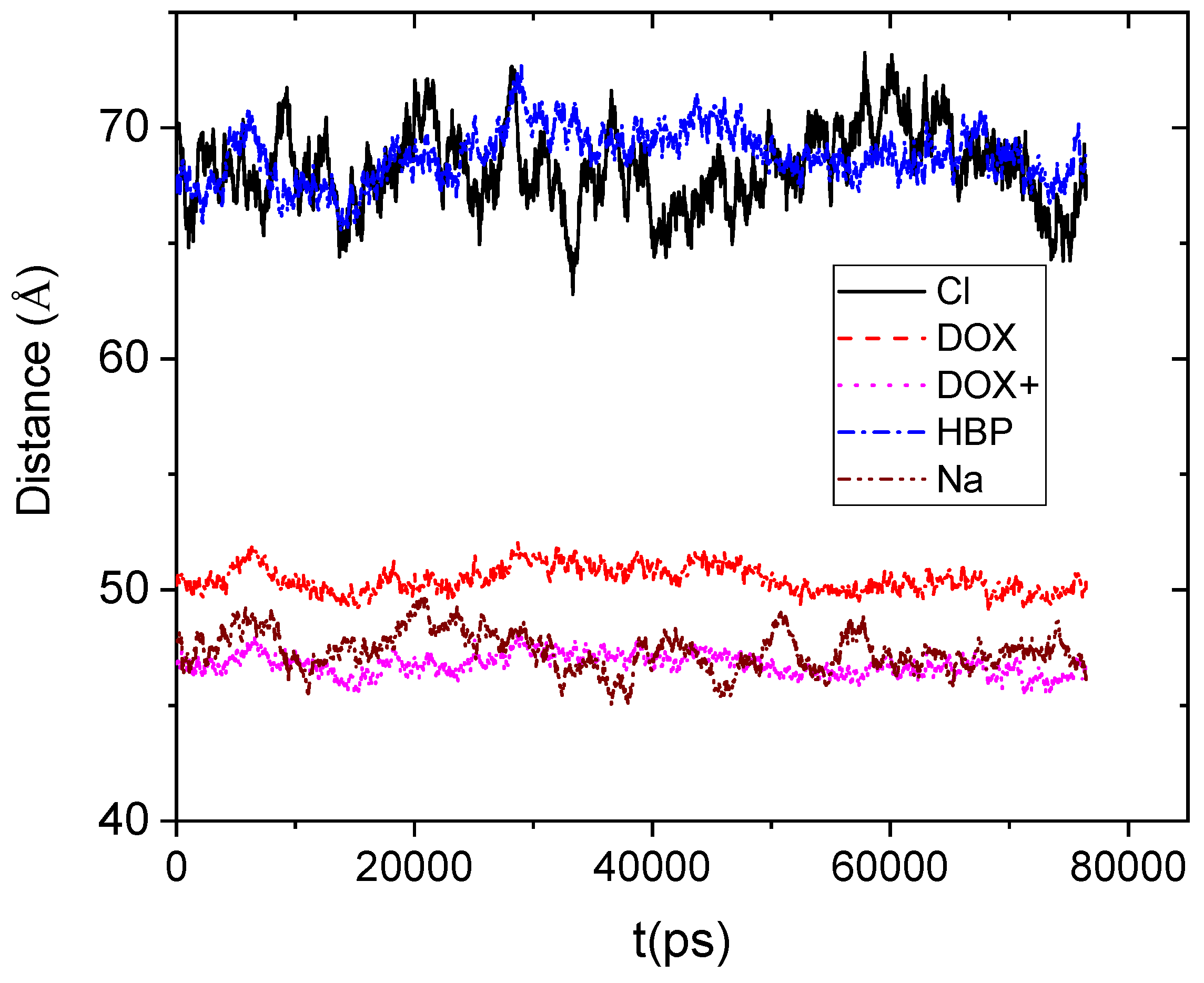
Figure 4 shows the average distance between the center of mass of the DPPG membrane and the center of mass of the other moieties.
If we take into account that the boundary of the HBP periphery was found to be at approximately 20 Å distance from its center of mass, it appears that drug molecules remain on average close to the HBP’s surface. However, the protonated doxorubicin molecules together with the Na+ counterions appear to be somewhat closer to the membrane. The closer proximity of the positively charged moieties should be attributed to the favorable Coulombic interactions with the membrane. Lack of such favorable electrostatic interactions between the DPPG membrane and the HBP, as well as the repulsive interactions with the Cl− counterions, can account for the larger distances between those moieties and the bilipid membrane.
To elaborate more on the spatial arrangement of the drug molecules with respect to the hyperbranched nanoparticle, we have calculated their mass distribution as a function of their distance from the center of mass of the HBP, as illustrated in
Figure 5.
The main feature characterizing the mass distributions of the drug molecules is their bimodal behavior. No such bimodal behavior for the protonated doxorubicin was noted in the absence of the membrane [
19]. The second peak of the drugs’ distribution located beyond the HBP boundary exhibits a partial overlap with the membrane distribution. The degree of this overlap is considerably higher between the distribution describing the protonated drug molecules and that of the membrane. Quantification of the degree of penetration of the drug molecules within the HBP’s structure and within the bilipid membrane can be made by calculating the area of the profiles within the boundaries of the HBP and the membrane respectively.
Table 2 and
Table 3 list the so-calculated degrees of drug penetration.
The percentages appearing in
Table 3 indicate that a significant amount of drug molecules have migrated from the HBP/drug complex towards the DPPG surface. This percentage is considerably higher for the protonated drug molecules, apparently due to the attractive Coulombic interactions with the negatively charged lipids. It must be noted here, that the percentages appearing in
Table 2 and
Table 3 reflect equilibrium conditions, since the timescale of the simulation is orders of magnitude longer compared to the timescale necessary for the drug molecules to reach the diffusive regime at conditions similar to the ones examined here [
19], while the average distances between the different molecules remain stable (see
Figure 4).
3.2. Charge Distributions
In our effort on one hand to discuss further the driving forces which are responsible for the mass distributions described before, and on the other hand to explore overcharging phenomena which are known to play a significant role in drug or gene-delivery formulations [
33,
34,
35] and in the conformational properties of the hyperbranched molecules [
36], we calculated component and overall charge distributions close to the HBP/drug complex and close to the membrane’s surface.
Figure 6 portrays the overall charge profile (arising from all components in the system) when moving radially outward with respect to the center of mass of the HBP (all charges henceforth are presented in units of an electron charge). It is shown that the overall charge profile exhibits a strong fluctuation around zero at distances extending from the center of mass of the PEGylated hyperbranched polyester to a region close to its periphery. At the outermost region, however, a net positive charge develops. As it was found, this charge arises from the positively charged drug molecules which are concentrated close to the HBP’s outer surface. Calculation of the overall charge of the complex rendered a net charge of approximately 8.5e (integration from the polymer’s center of mass to its boundary at ~22.5 Å).
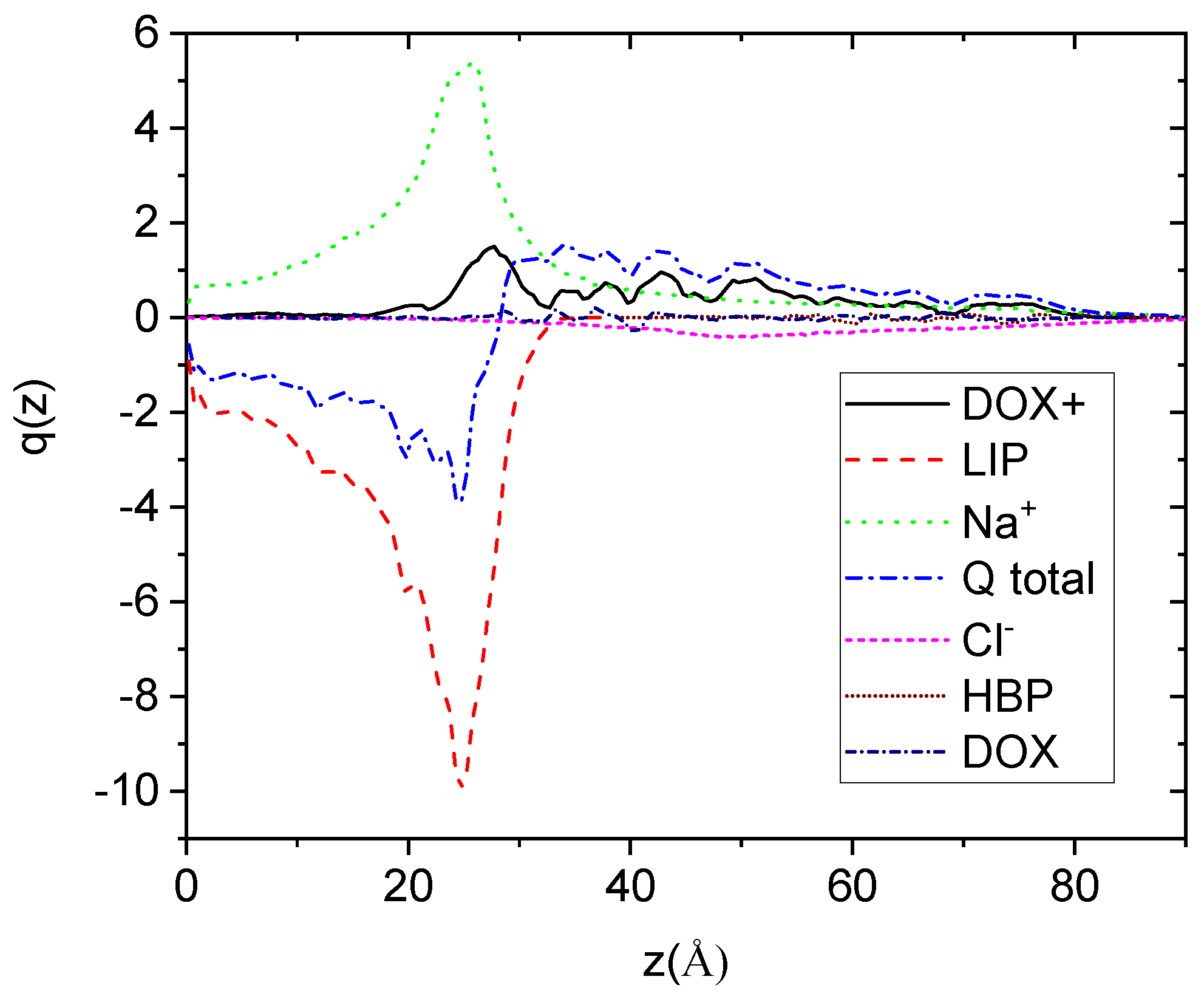
To explore the charge profiles with respect to the bilipid’s surface, we considered a reference frame with its origin in the center of mass of the bilayer and with the directions of its axes defined by the principal axes of inertia of the membrane. The profiles, shown in
Figure 7, were calculated along the direction normal to the plane defined by the directions of two of the principal axes of inertia which lie parallel to the membrane’s surface (here denoted as z).
Focusing on the overall sum of the distributions, it appears that there exists a net negative charge close to the membrane’s surface. The membrane boundary is located close to 30 Å as this has been determined independently by the corresponding mass distribution (not shown here). As anticipated, the net charge arises by the negatively charged lipids. The concentration of positive charges arising from the Na+ and the DOX+ drug molecules does not suffice to counterbalance the lipids’ negative charge. Estimation of the overall charge of the membrane complex results to a value of approximately −100 (integration from the membrane’s center of mass to its boundary in both directions), which is appreciably reduced (by about 22%) compared to the entire membrane charge (−128).
3.3. Effects of the Presence of the Membrane in the Clustering Behavior of Doxorubicin
As has been demonstrated in past studies (see Reference [
19] and references therein) Doxorubicin tends to self-associate in aqueous solutions forming oligomeric clusters. This behavior is attributed to hydrophobic forces and to the π−π electron interaction between the planar aromatic parts of the drug molecules. The formation of such drug aggregates and the structural characteristics of these clusters may be of paramount importance regarding their pharmacological efficiency, since they can affect their transport properties, their permeation through a membrane, and ultimately the final drug biodistribution. To monitor the changes imparted in the associative behavior of doxorubicin molecules in the presence of the polymeric host and the bilipid membrane, the relative arrangement of the drug molecules was examined through radial distribution functions arising from their centers of mass.
Figure 8 shows the radial distribution functions of the center of mass of the drug molecules (neutral and protonated) with and without the presence of the lipid membrane (the behavior without the presence of the membrane was taken from Reference [
19]). The distributions without the membrane are almost bimodal, characterized by a sharp peak close to an intermolecular distance of 5 Å and a second maximum close to 7–8 Å. The short-distance peak arises from the closest neighbors of a drug molecule and is consistent with the formation of dimers, while the peak corresponding to longer distances arises from the presence of neighboring clusters. It must be noted that in the case of the protonated drugs, the electrostatic repulsions due to the presence of the charged amine groups affect the spatial arrangement of the molecules (i.e., the relative intensity and the number of the peaks).
In the case of the protonated drugs in the presence of the membrane, it is observed that on average the separation between the formed clusters decreases (see the distance between the first and the second peak with and without the presence of the membrane). This can be associated with the electrostatic attraction of the protonated drug molecules by the membrane, which results in an enhancement of their tendency to cluster close to the lipid surface. This behavior could also be related to the effective higher concentration of the drug molecules in the system with the membrane. As far as the neutral drugs are concerned, a similar behavior can be observed. It is noticed that in the presence of the DPPG bilayer, the third peak arising from neighboring clusters exhibits a higher intensity compared to the analogous peak without the presence of the membrane. In other words, the limited diffusion of the drug clusters due to the geometric restriction imposed by the membrane, combined with hydrophobic forces, results in a higher degree of the drug molecules’ localization. In general, therefore, for both cases (protonated and neutral drugs), the presence of the bilipid membrane appears to reinforce the drug clustering behavior.
3.4. Hydrogen Bonding
In the absence of the DPPG membrane, it was found that apart from the geometric entrapment of the drug molecules within the polymeric structure, another mechanism, that of hydrogen bonding, was also responsible for the association of doxorubicin with the polymeric host [
19]. This route for complexation can play a key role in drug delivery processes [
37]. In the presence of the bilipid membrane, additional hydrogen-bonding-capable groups are introduced in the system, creating thus antagonistic to the HBP centers for the association of drug molecules. Recalling the image discussed in
Section 3.1 (
Figure 5 and
Table 3), it is of interest to examine the role of hydrogen bonding in the membrane/drug association. To this end, we have examined pair correlation functions of hydrogen-bonding-capable atoms. The criteria we followed for the identification of a hydrogen bond were based on the hydrogen-acceptor distance in combination with the angle formed by the donor-hydrogen-acceptor triplet. The maximum distance considered, was the extent of the corresponding peak in the pertinent pair correlation function, while the minimum donor-hydrogen-acceptor angle was taken to be 120° [
38,
39].
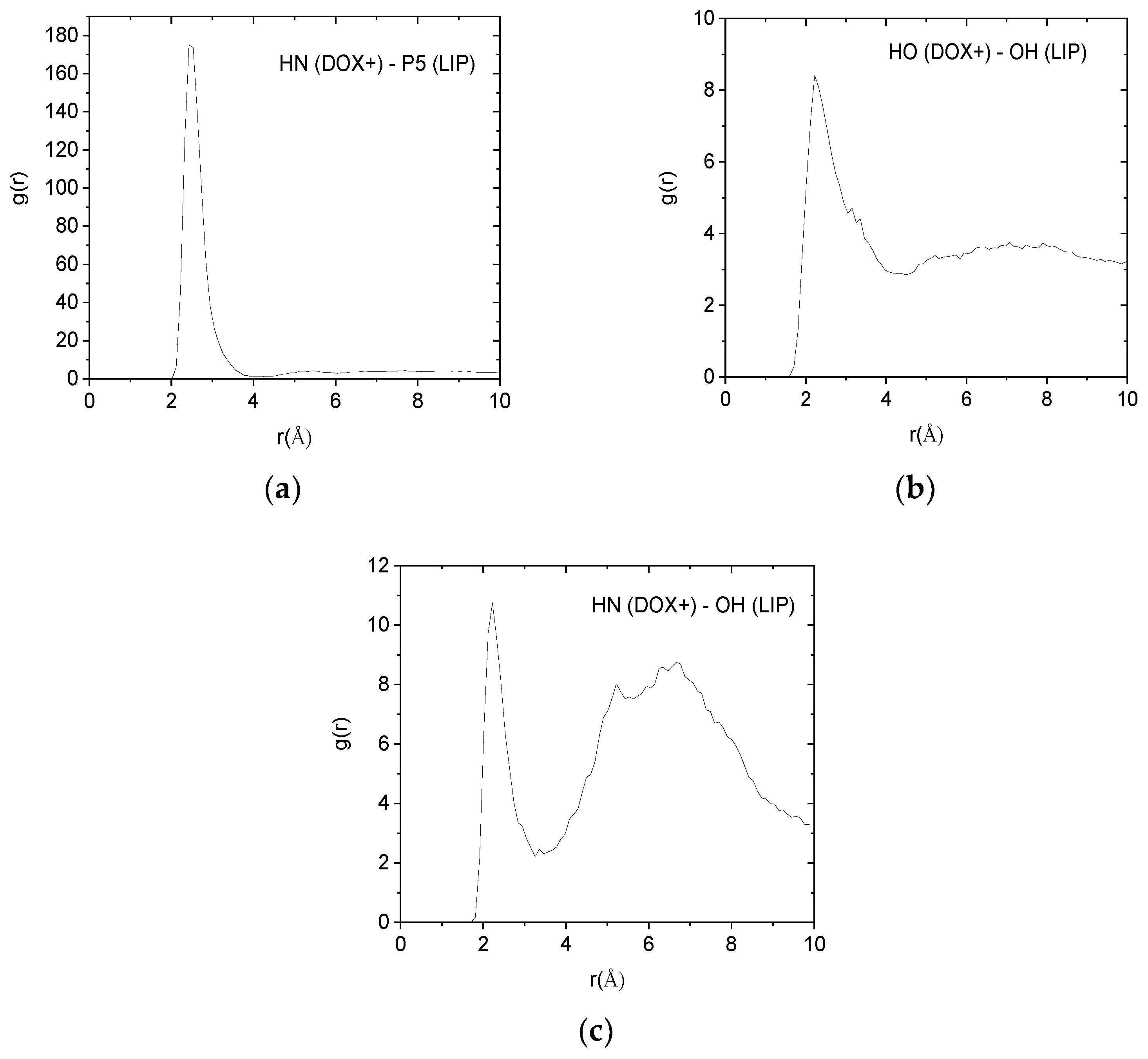
Such pair correlation functions between atoms belonging to the protonated doxorubicin molecules and to the lipids of the membranes are shown in
Figure 9. In all cases, the existence of a sharp peak close to 2.5 Å implies the presence of hydrogen bonds between the examined atomic pairs. The intensity of the hydrogen-bonding peak is indicative to the abundance of each kind of hydrogen bond. Therefore, the pair between the amine hydrogen of the DOX+ and the phosphorus atom of the lipid, appears to be the more abundant. Analogous pair correlation functions between the neutral drug molecules and the lipid are shown in
Figure 10.
To quantify the tendency for hydrogen-bond formation between the examined pairs, we have calculated the average number of each kind of hydrogen bond, per timestep, as presented in
Table 4.
Inspection of the values in
Table 4 verifies that hydrogen bonding between DOX+ amine hydrogens (HN) and lipid phosphorus atoms (P5) is the most frequent. A lower degree of hydrogen bonding which appears to be marginally statistically significant, is that of the DOX+ hydroxyl hydrogen (HO) and lipid hydroxyl oxygen (OH), while an even lower degree of hydrogen bonding is observed between the DOX+ amine hydrogen (HN) and the lipid’s hydroxyl oxygen (OH). In conjunction with the results from
Section 3.1 it can be concluded that one of the mechanisms which is responsible for the association between the protonated drug molecules and the membrane’s surface is the formation of hydrogen bonds (another mechanism is related to the electrostatic interactions, as discussed in
Section 3.2). As far as the neutral drugs are concerned, their association with the lipid membrane discussed in
Section 3.1 does not appear to be driven by hydrogen-bond formation (the average number of hydrogen bonds is not statistically significant). A more probable driving force for the latter is therefore the hydrophobic interactions of the neutral drug molecules with water, which can result in a more favorable interaction with the membrane’s surface.
3.5. Diffusional Behavior
The effects of the presence of the DPPG membrane in the clustering characteristics and in the associative behavior of the drug molecules are expected to be reflected in their transport properties as well. To obtain information regarding the diffusional motion of the different molecular components of the examined system, we monitored the mean squared displacement (MSD) of their center of mass as depicted in
Figure 11. For comparison purposes we have also included the diffusional behavior of the drug molecules in the membrane-free case [
19].
Focusing on the behavior of doxorubicin molecules, it appears that the presence of the DPPG membrane drastically affects the drug diffusional motion. Moreover, it appears to do so to a different degree depending on whether the drug molecules are neutral or protonated. The diffusional motion of the neutral drug at all the examined timescales is slower compared to the analogous behavior in the membrane-free system, remaining very close to that of the HBP. This observation can be rationalized on one hand by the fact that the majority of the neutral drug molecules are located within the HBP’s structure (see
Figure 5 and
Table 2) and on the other hand by the more restrictive environment (i.e., higher effective concentration compared to the membrane-free case, enhanced clustering behavior, partial association with the bilipid membrane) experienced by these molecules in the presence of the bilipid layer.
In contrast, the protonated drug molecules diffuse much faster compared to their behavior in the absence of the membrane as far as it concerns the short timescales. This trend changes at larger timescales where the transport of the center of mass of the latter becomes slower than the membrane-free case. The origin for the more complex behavior exhibited by the protonated drug should be related to the electrostatic interactions between the cationic doxorubicin molecules and the anionic part of the DPPG lipids. At short timescales, Coulombic interactions between the two oppositely charged moieties provide the driving force for an enhanced diffusive behavior. At larger timescales, where the effects of the more constrictive environment become more important, all those factors affecting the neutral drug molecules start affecting the clusters of the protonated drug molecules as well, resulting in the slowing down of their diffusional motion.
4. Conclusions
As was demonstrated by the analysis presented, the presence of the DPPG membrane imparts significant changes regarding the spatial distribution and the clustering behavior of the drug molecules. Almost 1/3 of the protonated and 1/5 of the neutral doxorubicin molecules appear to migrate from the drug/HBP complex towards the membrane’s surface. Localization of drug molecules close to the membrane’s surface enhances their tendency to form clusters, particularly for the positively charged drug molecules. The latter form persistent hydrogen bonds mainly with the phosphorus atoms of the hydrophilic part of the lipids. The affinity of the neutral doxorubicin molecules to the lipid bilayer appears to be driven by their hydrophobic nature. The uneven distribution of the DOX+ molecules and that of the counterions’ between the HBP and the bilipid membrane results in the development of positive and negative surface charge excesses, respectively. This may eventually lead to the coalescence of the HBP/drug complex with the membrane and thus towards a mechanism for a liposomal escape in formulations in which such HBP/doxorubicin complexes are encapsulated in a DPPG-based liposomal carrier.
The timescales associated with the migration of the neutral and the protonated doxorubicin molecules from the HBP/drug complex to the membrane’s surface, and thus the build-up of the final charge distribution, relates to the respective diffusional behavior. It appears that the presence of the DPPG membrane drastically affects the translational motion of doxorubicin with respect to the membrane-free case. The extent of the observed changes depends on the charge carried by the drug molecules. The neutral drug diffuses slower at all the examined timescales while the protonated drug diffuses faster at short timescales and slower at longer timescales, compared to the behavior observed in the absence of the DPPG bilayer.
The elementary mechanisms characterizing the interactions of doxorubicin molecules with a hyperbranched polymeric host in the presence of an anionic bilipid membrane may serve as a basis for a better understanding of the microscopic picture in other liposomal-based formulations for drug-delivery purposes, where similar interactions are expected to be present.
Author Contributions
Conceptualization, methodology, analysis software, writing—original draft preparation, writing—review and editing, supervision: K.K.; Data analysis, validation, visualization: P.A.; Interpretation of the results: K.K. and P.A.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Liu, Y.; Li, M.; Yang, Y.; Xia, Y.; Nieh, M.-P. The effects of temperature, salinity, concentration and PEGylated lipid on the spontaneous nanostructures of bicellar mixtures. Biochim. Biophys. Acta BBA Biomembr. 2014, 1838, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Ferber, D. GENE THERAPY: Safer and Virus-Free? Science 2001, 294, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Woodle, M.C.; Scaria, P. Cationic liposomes and nucleic acids. Curr. Opin. Colloid Interface Sci. 2001, 6, 78–84. [Google Scholar] [CrossRef]
- Kontogiannopoulos, K.N.; Assimopoulou, A.N.; Hatziantoniou, S.; Karatasos, K.; Demetzos, C.; Papageorgiou, V.P. Chimeric advanced drug delivery nano systems (chi-aDDnSs) for shikonin combining dendritic and liposomal technology. Int. J. Pharm. 2012, 422, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Tsermentseli, S.; Kontogiannopoulos, K.; Papageorgiou, V.; Assimopoulou, A. Comparative Study of PEGylated and Conventional Liposomes as Carriers for Shikonin. Fluids 2018, 3, 36. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef]
- Wenz, J.J.; Barrantes, F.J. Steroid Structural Requirements for Stabilizing or Disrupting Lipid Domains. Biochemistry 2003, 42, 14267–14276. [Google Scholar] [CrossRef]
- Tyteca, D.; Schanck, A.; Dufrêne, Y.F.; Deleu, M.; Courtoy, P.J.; Tulkens, P.M.; Mingeot-Leclercq, M.P. The macrolide antibiotic azithromycin interacts with lipids and affects membrane organization and fluidity: Studies on langmuir-blodgett monolayers, liposomes and J774 macrophages. J. Membr. Biol. 2003, 192, 203–215. [Google Scholar] [CrossRef]
- Li, J.; Ouyang, Y.; Kong, X.; Zhu, J.; Lu, D.; Liu, Z. A multi-scale molecular dynamics simulation of PMAL facilitated delivery of siRNA. RSC Adv. 2015, 5, 68227–68233. [Google Scholar] [CrossRef]
- Nademi, Y.; Amjad Iranagh, S.; Yousefpour, A.; Mousavi, S.; Modarress, H. Molecular dynamics simulations and free energy profile of Paracetamol in DPPC and DMPC lipid bilayers. J. Chem. Sci. 2014, 126, 637–647. [Google Scholar] [CrossRef]
- Kopeć, W.; Telenius, J.; Khandelia, H. Molecular dynamics simulations of the interactions of medicinal plant extracts and drugs with lipid bilayer membranes. FEBS J. 2013, 280, 2785–2805. [Google Scholar] [CrossRef]
- Antipina, A.Y.; Gurtovenko, A.A. Toward Understanding Liposome-Based siRNA Delivery Vectors: Atomic-Scale Insight into siRNA–Lipid Interactions. Langmuir 2018, 34, 8685–8693. [Google Scholar] [CrossRef] [PubMed]
- Tah, B.; Pal, P.; Mishra, S.; Talapatra, G.B. Interaction of insulin with anionic phospholipid (DPPG) vesicles. Phys. Chem. Chem. Phys. 2014, 16, 21657–21663. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, T.; Colherinhas, G.; de Oliveira Outi, F.; Fileti, E.E. Assessing the interaction between surfactant-like peptides and lipid membranes. RSC Adv. 2017, 7, 35973–35981. [Google Scholar] [CrossRef]
- Ergun, S.; Demir, P.; Uzbay, T.; Severcan, F. Agomelatine strongly interacts with zwitterionic DPPC and charged DPPG membranes. Biochim. Biophys. Acta BBA Biomembr. 2014, 1838, 2798–2806. [Google Scholar] [CrossRef]
- Habib, L.; Jraij, A.; Khreich, N.; Fessi, H.; Charcosset, C.; Greige-Gerges, H. Morphological and physicochemical characterization of liposomes loading cucurbitacin E, an anti-proliferative natural tetracyclic triterpene. Chem. Phys. Lipids 2014, 177, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Mourelatou, E.A.; Libster, D.; Nir, I.; Hatziantoniou, S.; Aserin, A.; Garti, N.; Demetzos, C. Type and Location of Interaction between Hyperbranched Polymers and Liposomes. Relevance to Design of a Potentially Advanced Drug Delivery Nanosystem (aDDnS). J. Phys. Cem. B 2011, 115, 3400–3408. [Google Scholar] [CrossRef]
- Oberoi, H.S.; Nukolova, N.V.; Kabanov, A.V.; Bronich, T.K. Nanocarriers for delivery of platinum anticancer drugs. Adv. Drug Deliv. Rev. 2013, 65, 1667–1685. [Google Scholar] [CrossRef]
- Karatasos, K. Self-Association and Complexation of the Anti-Cancer Drug Doxorubicin with PEGylated Hyperbranched Polyesters in an Aqueous Environment. J. Phys. Cem. B 2013, 117, 2564–2575. [Google Scholar] [CrossRef]
- Wang, Y.; Kong, W.; Song, Y.; Duan, Y.; Wang, L.; Steinhoff, G.; Kong, D.; Yu, Y. Polyamidoamine Dendrimers with a Modified Pentaerythritol Core Having High Efficiency and Low Cytotoxicity as Gene Carriers. Biomacromolecules 2009, 10, 617–622. [Google Scholar] [CrossRef]
- Jämbeck, J.P.M.; Lyubartsev, A.P. Another Piece of the Membrane Puzzle: Extending Slipids Further. J. Chem. Theory Comput. 2013, 9, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Jämbeck, J.P.M.; Lyubartsev, A.P. Derivation and Systematic Validation of a Refined All-Atom Force Field for Phosphatidylcholine Lipids. J. Phys. Cem. B 2012, 116, 3164–3179. [Google Scholar] [CrossRef] [PubMed]
- Jämbeck, J.P.M.; Lyubartsev, A.P. An Extension and Further Validation of an All-Atomistic Force Field for Biological Membranes. J. Chem. Theory Comput. 2012, 8, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA—An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Aided Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Rosso, L.; Betz, R.M.; Walker, R.C.; Gould, I.R. GAFFlipid: A General Amber Force Field for the accurate molecular dynamics simulation of phospholipid. Soft Matter 2012, 8, 9617–9627. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Darden, T.; Perera, L.; Li, L.; Pedersen, L. New tricks for modelers from the crystallography toolkit: The particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure 1999, 7, R55–R60. [Google Scholar] [CrossRef]
- Po, H.N.; Senozan, N.M. The Henderson-Hasselbalch Equation: Its History and Limitations. J. Chem. Educ. 2001, 78, 1499. [Google Scholar] [CrossRef]
- Dalmark, M.; Johansen, P. Molecular Association between Doxorubicin (Adriamycin) and DNA-Derived Bases, Nucleosides, Nucleotides, Other Aromatic Compounds, and Proteins in Aqueous Solution. Mol. Pharmacol. 1982, 22, 158–165. [Google Scholar] [PubMed]
- Ouyang, D.; Zhang, H.; Parekh, H.S.; Smith, S.C. Structure and Dynamics of Multiple Cationic Vectors−siRNA Complexation by All-Atomic Molecular Dynamics Simulations. J. Phys. Cem. B 2010, 114, 9231–9237. [Google Scholar] [CrossRef] [PubMed]
- Bielinska, A.U.; Chen, C.; Johnson, J.; Baker, J.R. DNA Complexing with Polyamidoamine Dendrimers: Implications for Transfection. Bioconjug. Chem. 1999, 10, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Fant, K.; Esbjorner, E.K.; Lincoln, P.; Norden, B. DNA condensation by PAMAM dendrimers: Self-assembly characteristics and effect on transcription. Biochemistry 2008, 47, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Lyulin, S.V.; Karatasos, K.; Darinskii, A.; Larin, S.; Lyulin, A.V. Structural effects in overcharging in complexes of hyperbranched polymers with linear polyelectrolytes. Soft Matter 2008, 4, 453–457. [Google Scholar] [CrossRef]
- Pignatello, R.; Musumeci, T.; Basile, L.; Carbone, C.; Puglisi, G. Biomembrane models and drug-biomembrane interaction studies: Involvement in drug design and development. J. Pharm. Bioallied Sci. 2011, 3, 4–14. [Google Scholar] [CrossRef]
- Lee, H.; Baker, J.R.; Larson, R.G. Molecular dynamics studies of the size, shape, and internal structure of 0% and 90% acetylated fifth-generation polyamidoamine dendrimers in water and methanol. J. Phys. Chem. B 2006, 110, 4014–4019. [Google Scholar] [CrossRef]
- Chiessi, E.; Cavalieri, F.; Paradossi, G. Water and Polymer Dynamics in Chemically Cross-Linked Hydrogels of Poly(vinyl alcohol): A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2007, 111, 2820–2827. [Google Scholar] [CrossRef]
Figure 1.
(a) A single dipalmitoylphosphatidylglycerol (DPPG) lipid. (b) Doxorubicin. (c) The structure of the PEGylated hyperbranched polyester. Each PEG arm consisted of 20 monomers.
Figure 1.
(a) A single dipalmitoylphosphatidylglycerol (DPPG) lipid. (b) Doxorubicin. (c) The structure of the PEGylated hyperbranched polyester. Each PEG arm consisted of 20 monomers.
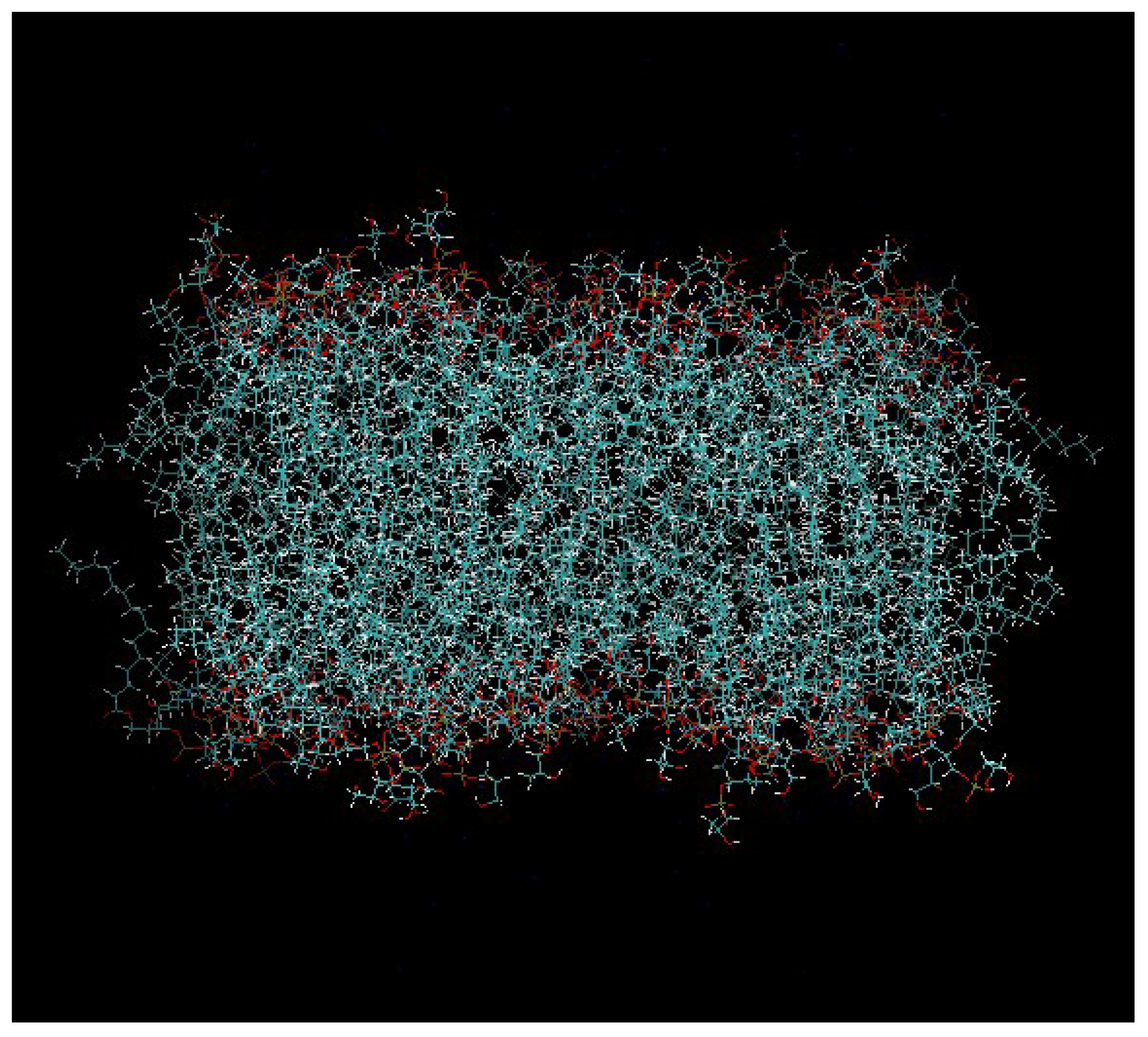
Figure 2.
The pre-equilibrated model of the DPPG membrane that was used in our study.
Figure 2.
The pre-equilibrated model of the DPPG membrane that was used in our study.
Figure 3.
(a) The constructed system including water molecules (shown in red-white). (b) The same system without the water molecules. Blue beads represent the chlorine ions and orange beads the sodium ions. Each drug molecule is shown in different color and in rod representation, while the PEGylated hyperbranched molecule is shown in red color. In the lower part of the snapshot, the lipids of the membrane are shown in wire representation.
Figure 3.
(a) The constructed system including water molecules (shown in red-white). (b) The same system without the water molecules. Blue beads represent the chlorine ions and orange beads the sodium ions. Each drug molecule is shown in different color and in rod representation, while the PEGylated hyperbranched molecule is shown in red color. In the lower part of the snapshot, the lipids of the membrane are shown in wire representation.
Figure 4.
Average distance as a function of time between the center of mass of the lipid membrane (considered as an individual molecule including all the lipids) and those of the hyperbranched polymer, the counterions (Cl−, Na+), and the neutral (DOX) and the protonated (DOX+) drug molecules.
Figure 4.
Average distance as a function of time between the center of mass of the lipid membrane (considered as an individual molecule including all the lipids) and those of the hyperbranched polymer, the counterions (Cl−, Na+), and the neutral (DOX) and the protonated (DOX+) drug molecules.
Figure 5.
Weight distribution of the drug molecules and of the other moieties with respect to the center of mass of the hyperbranched polymer (a spherical symmetry of the hyperbranched polyester (HBP) molecule has been considered). The notation in the legend is as in
Figure 4, with LIP denoting the DPPG membrane. The vertical dashed line denotes the location of the radius of gyration of the polymer.
Figure 5.
Weight distribution of the drug molecules and of the other moieties with respect to the center of mass of the hyperbranched polymer (a spherical symmetry of the hyperbranched polyester (HBP) molecule has been considered). The notation in the legend is as in
Figure 4, with LIP denoting the DPPG membrane. The vertical dashed line denotes the location of the radius of gyration of the polymer.
Figure 6.
Overall charge distribution with respect to the HBP center of mass arising from all the components in the system. The vertical dashed line denotes the surface boundary of the hyperbranched nanoparticle.
Figure 6.
Overall charge distribution with respect to the HBP center of mass arising from all the components in the system. The vertical dashed line denotes the surface boundary of the hyperbranched nanoparticle.
Figure 7.
Symmetrized charge distributions of all the moieties in the examined system, as a function of the distance from the plane, which is defined from the center of mass of the lipid membrane (considered as an individual molecule) and the two principal inertia axes, which are parallel to the lipid layer. “Q total” denotes the sum of all the component charge profiles.
Figure 7.
Symmetrized charge distributions of all the moieties in the examined system, as a function of the distance from the plane, which is defined from the center of mass of the lipid membrane (considered as an individual molecule) and the two principal inertia axes, which are parallel to the lipid layer. “Q total” denotes the sum of all the component charge profiles.
Figure 8.
Radial distribution functions of the center of mass of the doxorubicin molecules in the presence (solid lines) and in the absence (dashed lines) of the bilipid membrane, for the (a) neutral and the (b) protonated drug molecules.
Figure 8.
Radial distribution functions of the center of mass of the doxorubicin molecules in the presence (solid lines) and in the absence (dashed lines) of the bilipid membrane, for the (a) neutral and the (b) protonated drug molecules.
Figure 9.
Pair correlation functions among atoms belonging to the protonated drug molecules (DOX+) and the DPPG lipids (LIP). (a) Pairs between amine hydrogens of DOX+ (HN) and hydroxyl oxygens of the lipid (OH). (b) Pairs between hydroxyl hydrogens (HO) of DOX+ and hydroxyl oxygens (OH) of the lipid. (c) Pairs between amine hydrogens of DOX+ (HN) and phosphorus (P5) of the lipid.
Figure 9.
Pair correlation functions among atoms belonging to the protonated drug molecules (DOX+) and the DPPG lipids (LIP). (a) Pairs between amine hydrogens of DOX+ (HN) and hydroxyl oxygens of the lipid (OH). (b) Pairs between hydroxyl hydrogens (HO) of DOX+ and hydroxyl oxygens (OH) of the lipid. (c) Pairs between amine hydrogens of DOX+ (HN) and phosphorus (P5) of the lipid.
Figure 10.
Pair correlation functions among atoms belonging to the neutral drug molecules (DOX) and the DPPG lipids (LIP). (a) Pairs between amine hydrogens of DOX (HN) and hydroxyl oxygens of the lipid (OH). (b) Pairs between hydroxyl hydrogens (HO) of DOX and hydroxyl oxygens (OH) of the lipid. (c) Pairs between amine hydrogens of DOX (HN) and phosphorus (P5) of the lipid.
Figure 10.
Pair correlation functions among atoms belonging to the neutral drug molecules (DOX) and the DPPG lipids (LIP). (a) Pairs between amine hydrogens of DOX (HN) and hydroxyl oxygens of the lipid (OH). (b) Pairs between hydroxyl hydrogens (HO) of DOX and hydroxyl oxygens (OH) of the lipid. (c) Pairs between amine hydrogens of DOX (HN) and phosphorus (P5) of the lipid.
Figure 11.
Mean square displacement arising from the center of mass of the drug molecules, the hyperbranched polyester, and the bilipid membrane (considered as a single moiety including all the lipids). The behavior of the drug molecules in the membrane-free case is also shown for comparison purposes.
Figure 11.
Mean square displacement arising from the center of mass of the drug molecules, the hyperbranched polyester, and the bilipid membrane (considered as a single moiety including all the lipids). The behavior of the drug molecules in the membrane-free case is also shown for comparison purposes.
Table 1.
Number of molecules/ions of each type included in the simulated system.
Table 1.
Number of molecules/ions of each type included in the simulated system.
| PEGylated Hyperbranched Polymer | DPPG Lipids | Neutral Drug Molecules | Positively Charged Drugs | Water | Chlorine Ions | Sodium Ions |
|---|
| 1 | 128 | 19 | 31 | 54234 | 35 | 132 |
Table 2.
Average number and degree of penetration of drug molecules within the HBP’s structure.
Table 2.
Average number and degree of penetration of drug molecules within the HBP’s structure.
| Molecular Species | Number of Molecules | Percentage (%) |
|---|
| DOX | 8 | 42 |
| DOX+ | 11 | 35 |
Table 3.
Average number and degree of penetration of drug molecules within the membrane’s structure.
Table 3.
Average number and degree of penetration of drug molecules within the membrane’s structure.
| Molecular Species | Number of Molecules | Percentage (%) |
|---|
| DOX | 3.6 | 19 |
| DOX+ | 10 | 30 |
Table 4.
Average number of hydrogen bonds per timestep for the examined pairs.
Table 4.
Average number of hydrogen bonds per timestep for the examined pairs.
| Drug/LIP | HN (Drug)-OH(LIP) | HN (Drug)-P5(LIP) | HO (Drug)-OH(LIP) |
|---|
| DOX+/LIP | 1.6 ± 1.1 | 10.1 ± 2.2 | 2.5 ± 1.5 |
| DOX/LIP | 0.5 ± 0.8 | 0.0 ± 0.2 | 0.8 ± 0.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}