Abstract
A numerical and experimental investigation in to the role of gasoline surrogates and their reduced chemical kinetic mechanisms in spark ignition (SI) engine knocking has been carried out. In order to predict autoignition of gasoline in a spark ignition engine three reduced chemical kinetic mechanisms have been coupled with quasi-dimensional thermodynamic modelling approach. The modelling was supported by measurements of the knocking tendencies of three fuels of very different compositions yet an equivalent Research Octane Number (RON) of 90 (ULG90, PRF90 and 71.5% by volume toluene blended with n-heptane) as well as iso-octane. The experimental knock onsets provided a benchmark for the chemical kinetic predictions of autoignition and also highlighted the limitations of characterisation of the knock resistance of a gasoline in terms of the Research and Motoring octane numbers and the role of these parameters in surrogate formulation. Two approaches used to optimise the surrogate composition have been discussed and possible surrogates for ULG90 have been formulated and numerically studied. A discussion has also been made on the various surrogates from the literature which have been tested in shock tube and rapid compression machines for their autoignition times and are a source of chemical kinetic mechanism validation. The differences in the knock onsets of the tested fuels have been explained by modelling their reactivity using semi-detailed chemical kinetics. Through this work, the weaknesses and challenges of autoignition modelling in SI engines through gasoline surrogate chemical kinetics have been highlighted. Adequacy of a surrogate in simulating the autoignition behaviour of gasoline has also been investigated as it is more important for the surrogate to have the same reactivity as the gasoline at all engine relevant conditions than having the same RON and Motored Octane Number (MON).
1. Introduction
The knock phenomenon limits the compression ratio and the thermodynamic efficiency of a spark ignition (SI) engine. At some operating conditions, avoiding it will force the spark advance away from the maximum brake torque point further lowering engine performance. If knock occurs, it can be severely damaging to the engine components. Knock, i.e., onset of strong pressure oscillations, has been known to be caused by the autoignition of the end gas ahead of the propagating flame which is controlled by chemical kinetics. Therefore, numerical modelling of autoignition may be used to predict knock onset. This has been demonstrated in various studies by modelling the autoignition chemistry of a simpler gasoline surrogate such as primary reference fuels (PRFs), i.e., a mixture of iso-octane and n-heptane [1,2,3].
Chemical kinetic mechanisms are routinely validated against shock tube and rapid compression machine measurements of the ignition delay times () of various gasoline surrogate fuels. The conditions for these laboratory measurements are similar to those which prevail before ignition in homogeneous charge compression ignition (HCCI) or controlled autoignition (CAI) engine, and the recent kinetic mechanisms, e.g., [4,5,6], perform remarkably well in these regimes. Application of such gasoline surrogate mechanisms have been seen in various studies demonstrating autoignition predictions in HCCI and CAI engines, [4,7]. The question arises of whether the reduced chemical mechanisms will perform equally well in predicting autoignition in SI engines. This work attempts to address this subject.
2. Practical Gasoline Surrogates
The simplest surrogate, a primary reference fuel (PRF), i.e., mixture of iso-octane and n-heptane, is used in the well known Research and Motored Octane Number (RON and MON) tests to quantify the knock resistance of a gasoline by matching knock intensities in a standard engine. Compared to the RON test, in the MON test the end gas temperature is higher at the same pressures and therefore, unless gasoline exhibits a negative temperature coefficient (NTC) phase at those temperatures, the MON value tends to be lower than RON. The sensitivity S, i.e., the difference of octane numbers () is a measure of fuel’s response to varying pressures and temperatures. PRF’s have zero sensitivity by definition. However, gasoline, owing to its complex composition, is likely to exhibit autoignition behaviour different from that of the PRF which matches its RON or the MON value, when the pressure and temperature history of the end gas does not match the standard RON and MON tests. Blends of iso-octane, n-heptane with toluene (Toluene Reference Fuels, TRFs) have an advantage of having non-zero sensitivity. Binary blends of toluene with iso-octane and n-heptane have also been studied in order to isolate their cross-oxidation chemistry [8], however such blends have found little application as gasoline surrogates.
Use of iso-octane and n-heptane as components of a surrogate is hardly avoidable as they represent linear and branched alkanes, major gasoline components. Besides, the chemistry leading to their auto-ignition is relatively well understood. However, gasoline does not consists only of alkanes. Thus, EN228, the European standard for gasoline specifies aromatic content of up to 35% by volume, it also allows 5% oxygenates by volume. Therefore, it seems natural to seek surrogates going beyond PRF’s and TRF’s which contain compounds approximating various families of hydrocarbons present in the gasoline [9].
Table 1 presents a list of such gasoline surrogates for which the auto-igntion delay times were experimentally determined using rapid compression machines (RCM), shock tubes (ST) and HCCI engines. The work of Gauthier et al. [10] presents a fairly comprehensive shock tube study of two TRFs for conditions of 12–25 and 45–60 atm at 850–1280 K. Two TRF’s were proposed to approximate a standard research gasoline, RD387, with an anti knock index (AKI) of 87. The measurements demonstrated a good similarity between the surrogates and the gasoline. However the RON and MON of these surrogates were later determined by Knop et al. [11] and the AKI of the surrogates were found to be lower than 87, see Table 1. But, even more importantly, the sensitivity S of the two TRFs was found to be much lower than the usual gasoline range of S of 8 to 12 points.

Table 1.
Volumetric composition of various gasoline surrogate blends the iginition delays of which have been measured.
While shock tube measurements offer an important benchmark for validation of the chemical kinetic mechanisms, they are difficult to perform at temperatures below 850 K and this makes validation of chemical kinetic mechanisms at engine conditions difficult. At these temperatures, many individual components of gasolines have ignition delays decreasing with temperature, the phenomenon known as negative temperature coefficient (NTC) behaviour. Work of Mehl et al. [12] demonstrated that the sensitivity of a surrogate can be correlated to the slope of the NTC region, , while the values of the auto-ignition delay time in the NTC region depend on the AKI of the surrogate. These two correlations and knowledge of the gasoline composition provides constraints for the aromatic and olefinic content of the surrogate which are key to achieving a realistic NTC behaviour and thus sensitivity. The PRF content was then varied to achieve the correct H/C ratio and the octane numbers. This approach was applied to another RD387 gasoline [12], for which a 4-component surrogate (Table 1, last entry) was proposed. Work of Kukkadapu et al. [13] further investigated one of the TRF formulation proposed in [10]; this surrogate is referred to as Gauthier TRF-A (iso-octane 63%, n-heptane 17%, toluene 20% by volume). Comparison of igntion delays for Gauthier TRF-A and the 4-component surrogate proposed by Mehl et al. [12], measured in an RCM, Kukkadapu et al. [13] revealed that the correlations of Mehl et al. [12] overestimate the fuel sensitivity thereby indicating a weakness of an empirical prediction of octane numbers.
Ethanol addition to gasoline is now common mainly due to legislative impetus. Ethanol acts as an octane improver and is commonly blended in amounts of up to 10% by volume. Works of Fikri et al. [14] and Cancino et al. [15] performed shock tube measurements of four multicomponent gasoline surrogates including ethanol, see Table 1.
The EN 228 gasoline standard specifies a maximum olefin content of 18% vol. Most European gasolines have olefin content between 5% and 9% vol, mostly branched rather than straight or cyclic compounds [9]. Oxidation characteristics of some olefins, such as 1-hexene, cyclohexene and 1-pentene have been studied, however, other very common ones, e.g., 2-methyl-2-butene are little studied. It is because of this lack of understanding of common gasoline olefins that proposed chemical kinetic schemes differ significantly in olefine oxidation pathways. For autoignition simulations, the choice of olefin is crucial for producing correct ignition delays while also matching the H/C ratio. Surrogates tabulated in Table 1 have therefore only limited capability to emulate gasoline in SI engine conditions.
3. Chemical Kinetics Schemes for Gasoline Surrogates
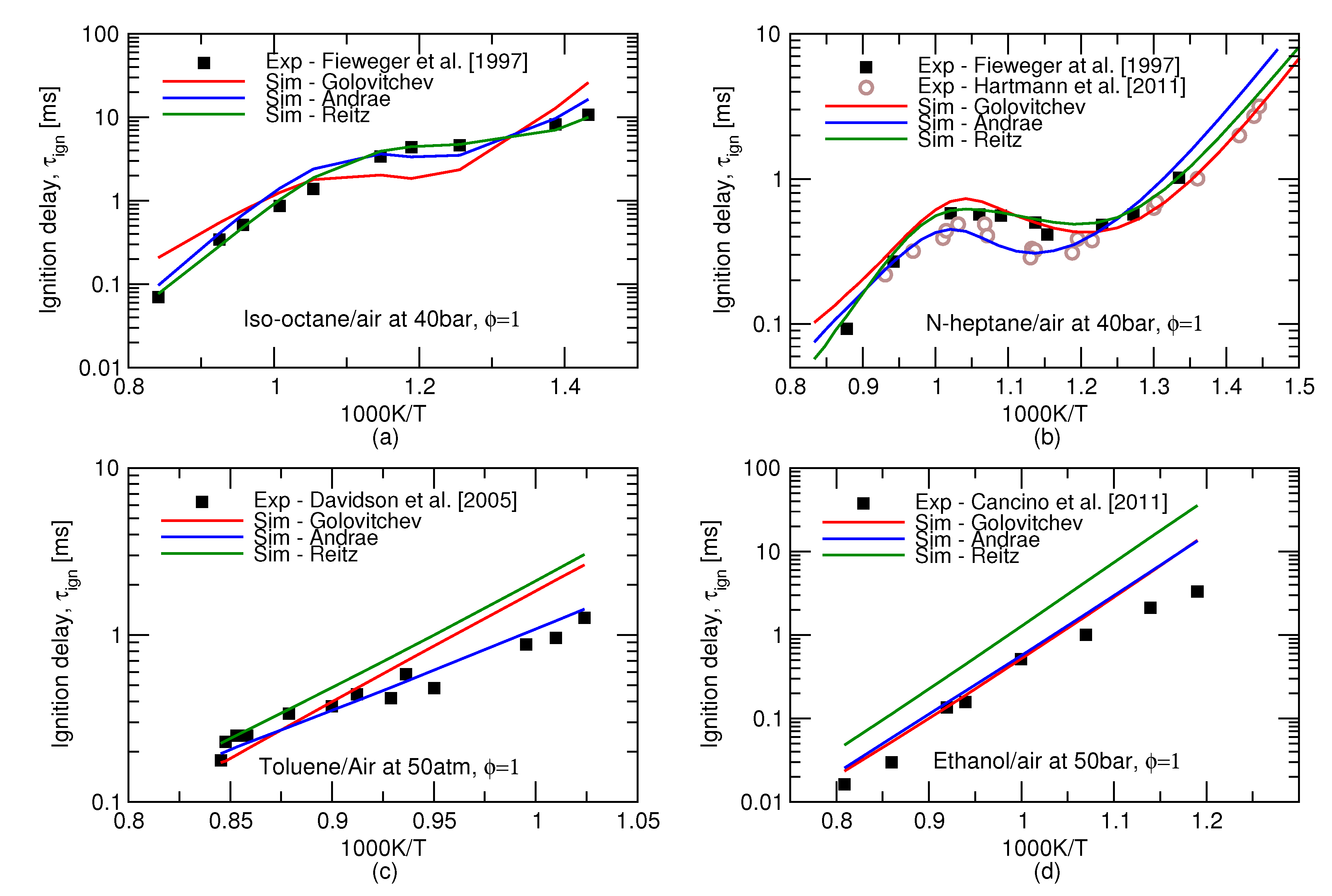
An earlier study [16] compared autoignition predictions in an adiabatic homogeneous reactor and an HCCI engine using 8 chemical kinetics mechanisms of different sizes. However, these mechanisms can describe only a limited amount of gasoline compounds, this is why this work turns to more comprehensive, semi-detailed kinetic schemes referred to as Andrae’s [4], Golovitchev’s [5], and Reitz’ [6], mechanisms. For individual substances, all these three mechanisms are capable of producing ignition delay times in fairly good agreement to experiments, see Figure 1. It may be inferred from this Figure that biggest discrepancy between experiments and models arises at high pressures and low, 600–850 K, temperatures. The observed difference would amount to a substantial deviation in terms of crank angle timing, e.g., at 1500 rpm, 1 ms equals 9 of the crank rotation. The integrated error along the complete history of the end gas may therefore result in a substantial difference between the predicted and observed autoignition onsets.
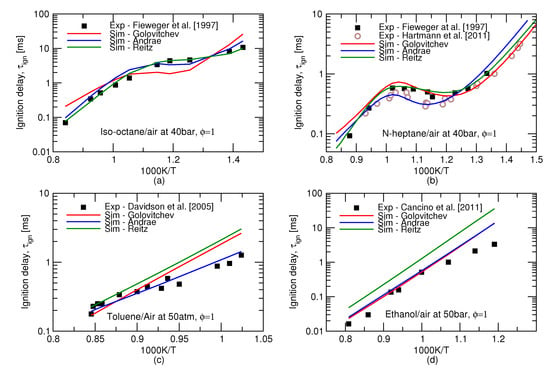
Figure 1.
Ignition delay time predictions (lines) of four individual key gasoline surrogate components: (a) iso-octane, (b) n-heptane, (c) toluene (d) ethanol. Symbols show shock tube measurements of Fieweger et al. [17], Hartmann et al. [18], Davidson et al. [19] and Cancino et al. [15].
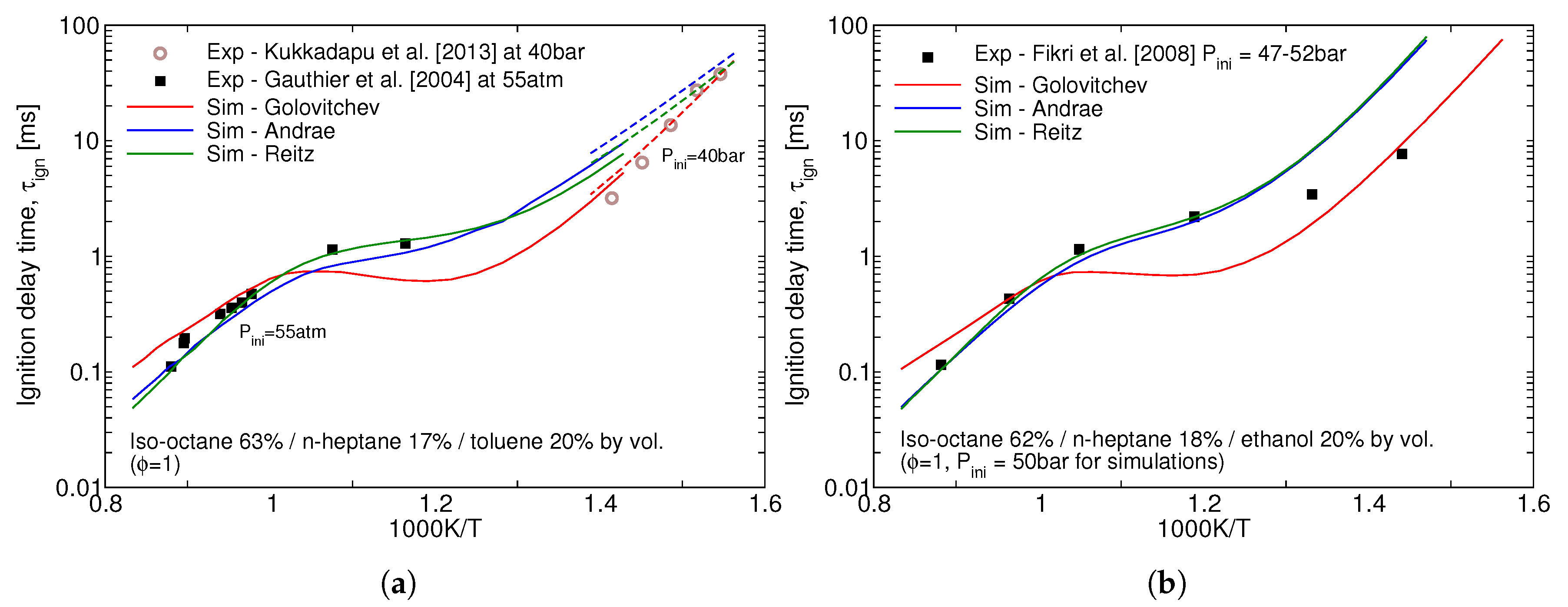
Performance of the three reduced mechanisms studied in this work for the ignition delay time predictions for the gasoline surrogates remains similar to the one for the individual icomponents. Ignition delay times for the stoichiometric mixtures of two surrogates at high pressures is presented in Figure 2. The over-prediction of the NTC behaviour of iso-octane by Golovitchev model as seen in Figure 1a is inheriteded in the surrogate simulations as well and, as a consequence, considerably lower ignition delay times are produced for both surrogates. It can be seen that Andrae’s mechanism outperforms the others at low, 600–850 K, temperatures crucial for the prediction of autoignition in SI engines.
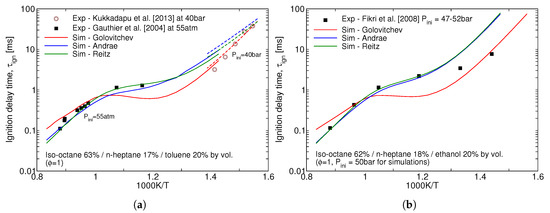
Figure 2.
Predicted vs. measured ignition delay times of the (a) Gauthier TRF-A and (b) an ethanol-containing gasoline surrogate by Fikri et al. [14].
The chemical kinetic calculations presented in this work have been done using a solver routine written in Fortran as part of the library of 1-D engine modelling code at Leeds University, refered to as LUSIE (Leeds University Spark Ignition Engine). For chemical kinetics, the solution of the stiff-type differential equations is carried out by using an implementation of the modified extended backward differentiation formulas developed by Cash [20]. The chemistry solver has been coupled with the engine simulation package in which the main combustion event is modelled using a multi-zone thermodymanic approach to flame propagation [21].
4. Gasoline Surrogate Formulation
Optimal surrogate formulation depends on its application. A large number of physical and chemical properties of the target fuel can be targeted in the surrogate, among those are the distillation curve, RON, MON, stoichiometric air-to-fuel ratio, molecular weight, thermal conductivity and laminar burning velocity. At the same time, the number of components in gasoline surrogate is limited by the availability of chemical kinetic data and complexity of blending rules. Since gasoline constituents belong to one of the five main classes it seems natural to seek surrogate with the same number of components. One mathematical constraint which must always be met while determining the composition of a surrogate is that the sum of the mole or volume fractions of its constituents must be one. This means that for a n component surrogate, properties can be used as constraints to optimise the surrogate composition. For a correct prediction of the cumulative heat release the stoichiometric air to fuel ratio () and the calorific value of the gasoline must also be matched. The correct H/C/O atomic proportions and the molar mass, M will automatically produce the desired .
For the quasi-dimensional combustion modelling in SI engine coupled with chemical kinetic modelling of surrogate autoignition in the end gas, the H/C and O/C ratios are crucial. Therefore, these constraints are used in the determination of the surrogate composition:
Reproducing the autoignition behaviour of the gasoline is the biggest challenge in surrogate formulation. The capablity of the surrogate to represent the anti-knock properties of the petrol is only partially dependent on the used model for octane number. A true surrogate for gasoline autoignition will match ignition delay of the gasoline at all conditions. It will show the emergence of similar ignition precursors at similar rates to that of the gasoline and therefore it will have, not only the same RON and MON as the gasoline, but similar octane index no matter which engine the two are compared in. Matching just the RON and MON of the surrogate with that of the gasoline does not guarantee that the surrogate will reproduce the autoignition behaviour of the gasoline universally in all engines. To make matters worse, the empirical/theoretical octane number models are far from perfect.
Detailed composition-based octane number models such as [22] account for the non-linear blending interactions of surrogate constituents. The model developed in [22] accounts for the paraffin-olefin and paraffin-naphthene interactions. The non-linear octane blending between ethanol and other gasoline constituents is a subject of on-going research [23] but as yet no well tested octane number model has emerged. Pera and Knop [9] advocated the use of a linear-by-moles additivity rule for the TRF blends shown to perform better than the non-linear model of Morgan et al. [24] or the composition-based octane model of Ghosh et al. [22]. Pera and Knop [9] proposed an improvement to the linear-by-moles expression for TRFs by suggesting blend octane numbers for toluene (RON 116/MON 101.8) and demonstrated that their expression yielded the lowest absolute errors in comparison to 7 other octane number models [11]. Their approach has been found to produce appreciable octane numbers in the present work and due to its accuracy and simplicity it has been adopted in this work for the calculation of TRF octane numbers as well as surrogates containing olefin:
Equations (1) and (2) and the unity sum of molar fractions provide 5 constraints for the determination of the so-called properties-based surrogate. Alternative to this would be a composition-based surrogate formulated by representing major constituents of gasoline by a surrogate of that particular family. The resulting surrogate will be expected to have different properties from that of the target gasoline as the surrogate components do not correctly represent all of the substances in that family. However, one of the findings of this work is that a composition-based surrogate whose composition represents accurately fractions of the actual gasoline may perform superior in replicating the autoignition behaviour as compared with a purely properties based surrogate which may contain unrealistic amounts of aromatics and oxygenates.
5. Supporting Experiments
Experiments were performed in a single cylinder optical engine (LUPOE2-D) at Leeds University with a disc shaped combustion chamber, the details of the engine and the experimental methods has been presented in [25]. Iso-octane, as well as three fuels of very different compositions, commercial unleaded petrol ULG90, PRF and TRF, with the same RON of 90 were tested in the knocking regime to assess the differences in their auto-ignition behaviour. For all fuels stoichiometric mixtures were tested. TRF90 was blended with toluene and n-heptane only as most of the octane quality of ULG90 came from its branched paraffin content.
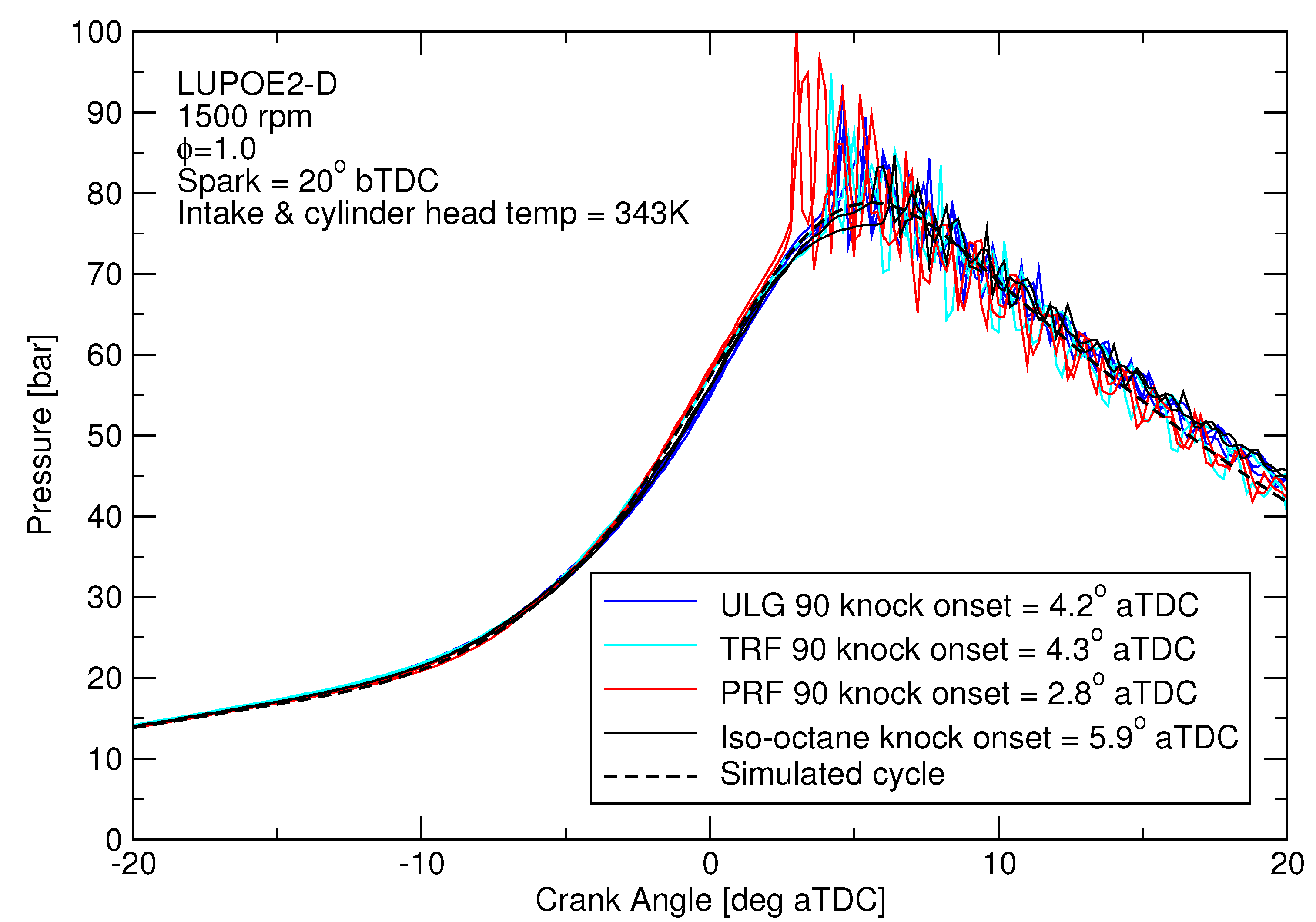
For autoignition modelling, cycles with similar pressure traces were chosen for the different fuels to allow a comparison of the knocking tendencies of different fuels at similar conditions, Figure 3. The subject of autoignition modelling with predictive combustion for the full range of slow and fast burning cycles has been covered in [1,2,3]. For this study, because fuel composition has only a small effect on the heat loss during compression phase, matching the cycle pressure effectively means very similar end-gas temperatures will result.
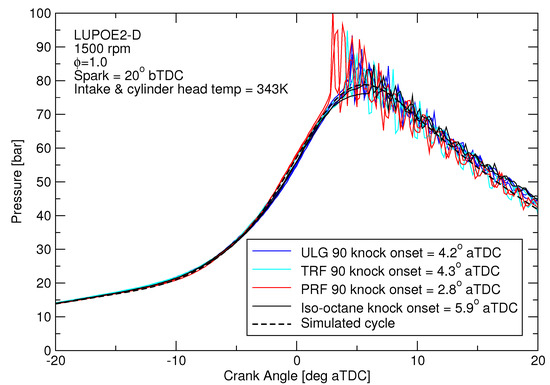
Figure 3.
The knocking experimental cycles for the four different fuels which have pressures similar to the pre-knock pressure of ULG90.
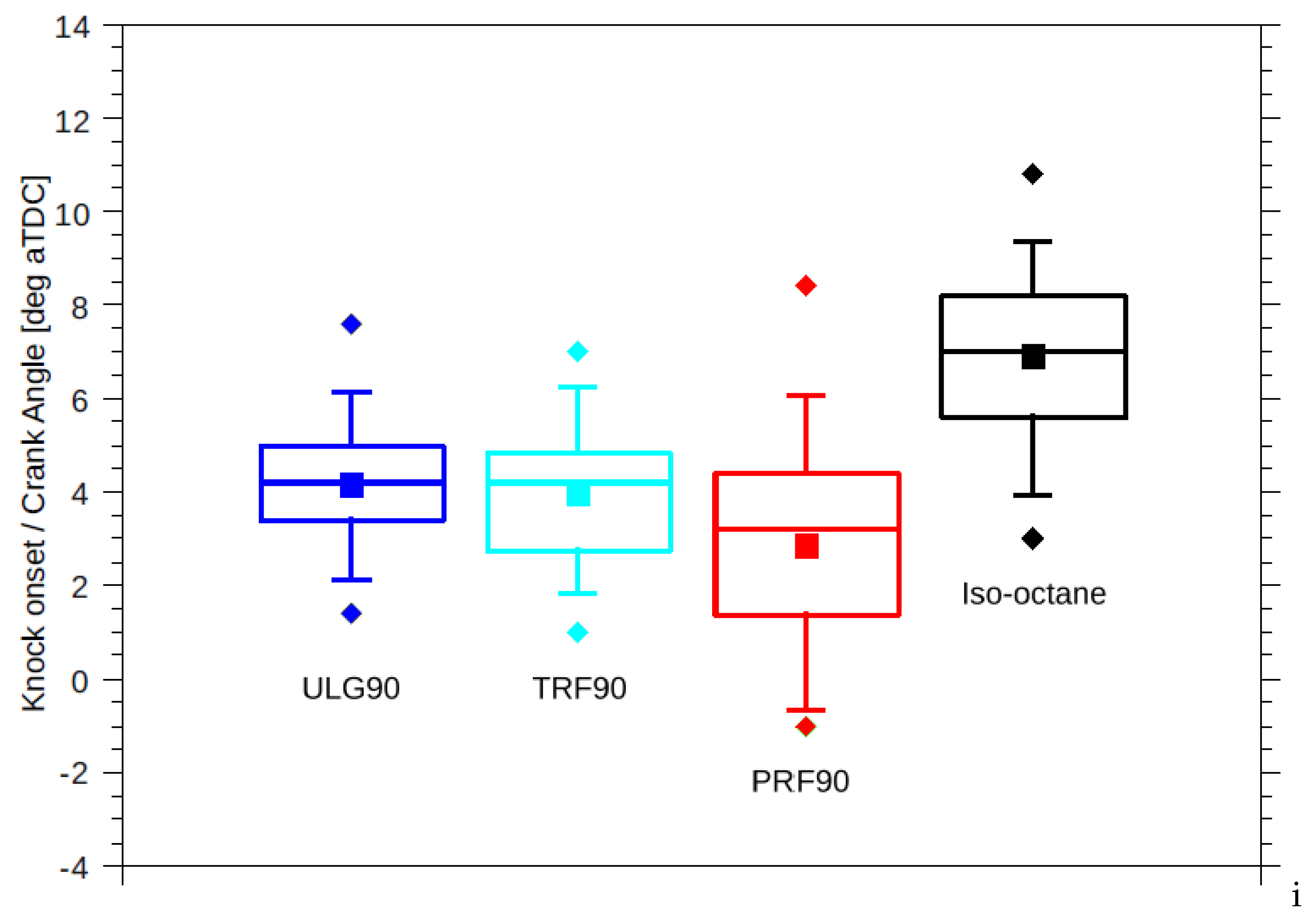
Difference in the reactivity of the two fuels was expected to manifest as different knock onsets regardless of the equivalent RON. However, TRF90 resulted in similar knock onset as ULG90 unlike PRF90 (Figure 4). The effective octane rating of ULG90 and TRF90 appears to be superior than the corresponding PRF of ON 90.
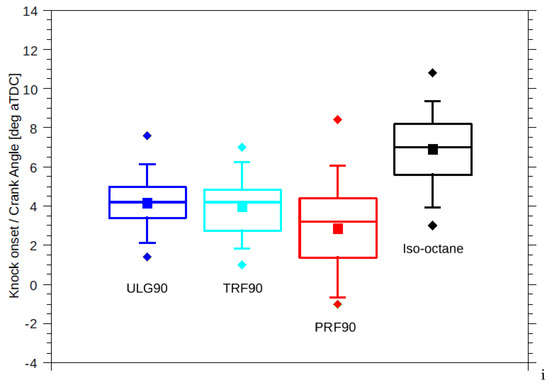
Figure 4.
Statistical variation in measured knock onsets for 100 combustion cycles of the four fuels tested. Squares: mean knock onset, diamonds: earlier and latest knock onsets, box: 25/75 percentile, whiskers: 5/95 percentile.
6. Results and Discussion
The relationship between the end gas thermodynamic state and the autoignitive tendency of a fuel can be depicted as ignition delay time () contours on a carpet plot as shown in Figure 5, Figure 6 and Figure 7.
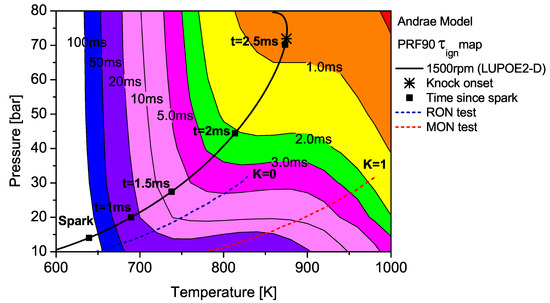
Figure 5.
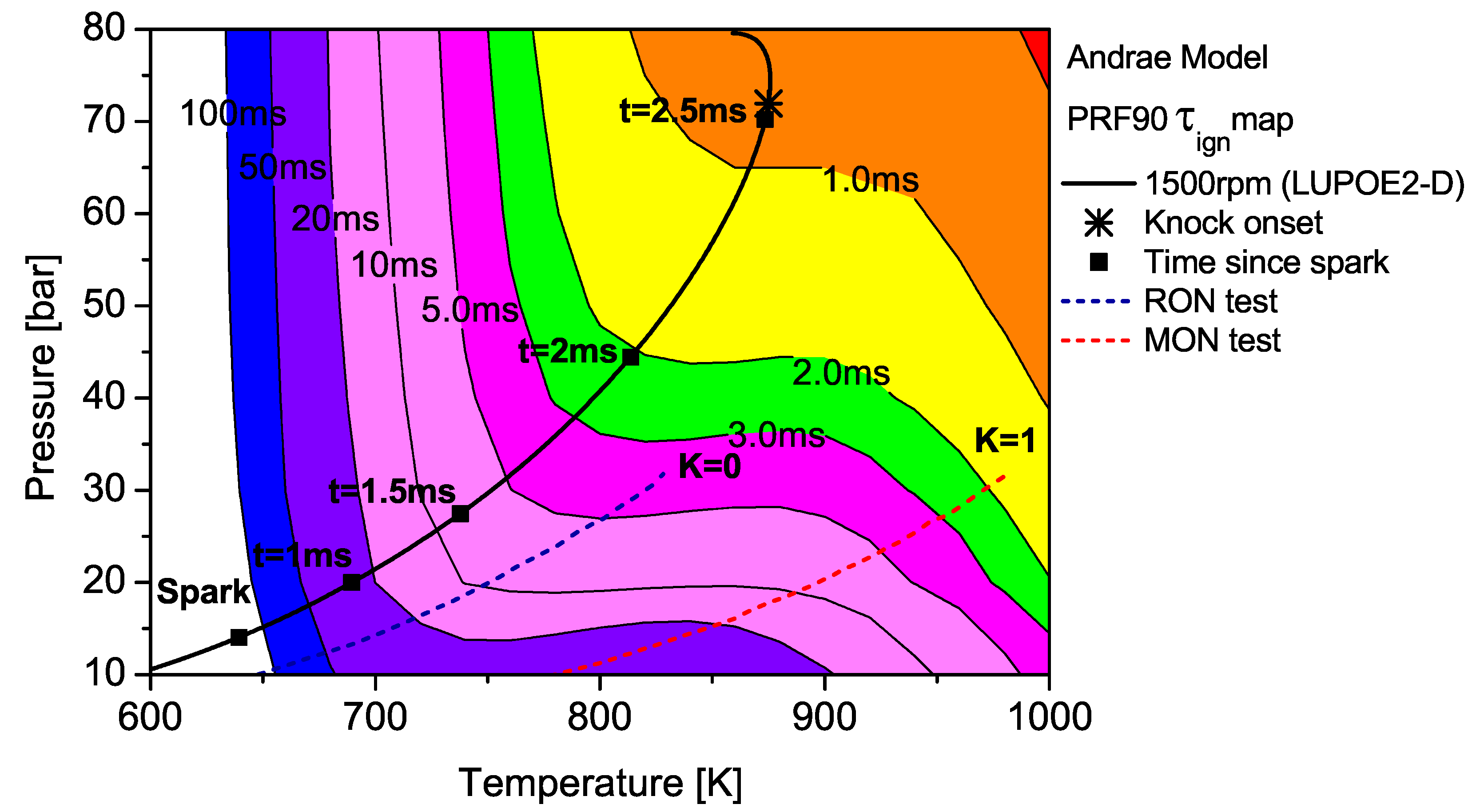
The contour map of PRF90 as predicted by the Andrae model superimposed with the unburned zone history of LUPOE2-D and typical RON/MON tests.
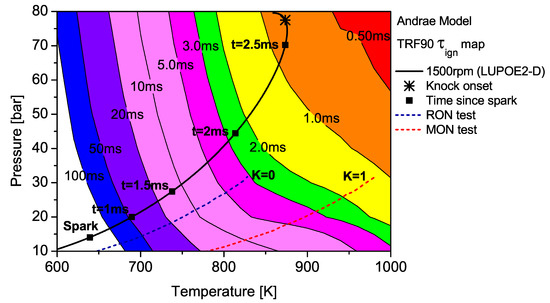
Figure 6.
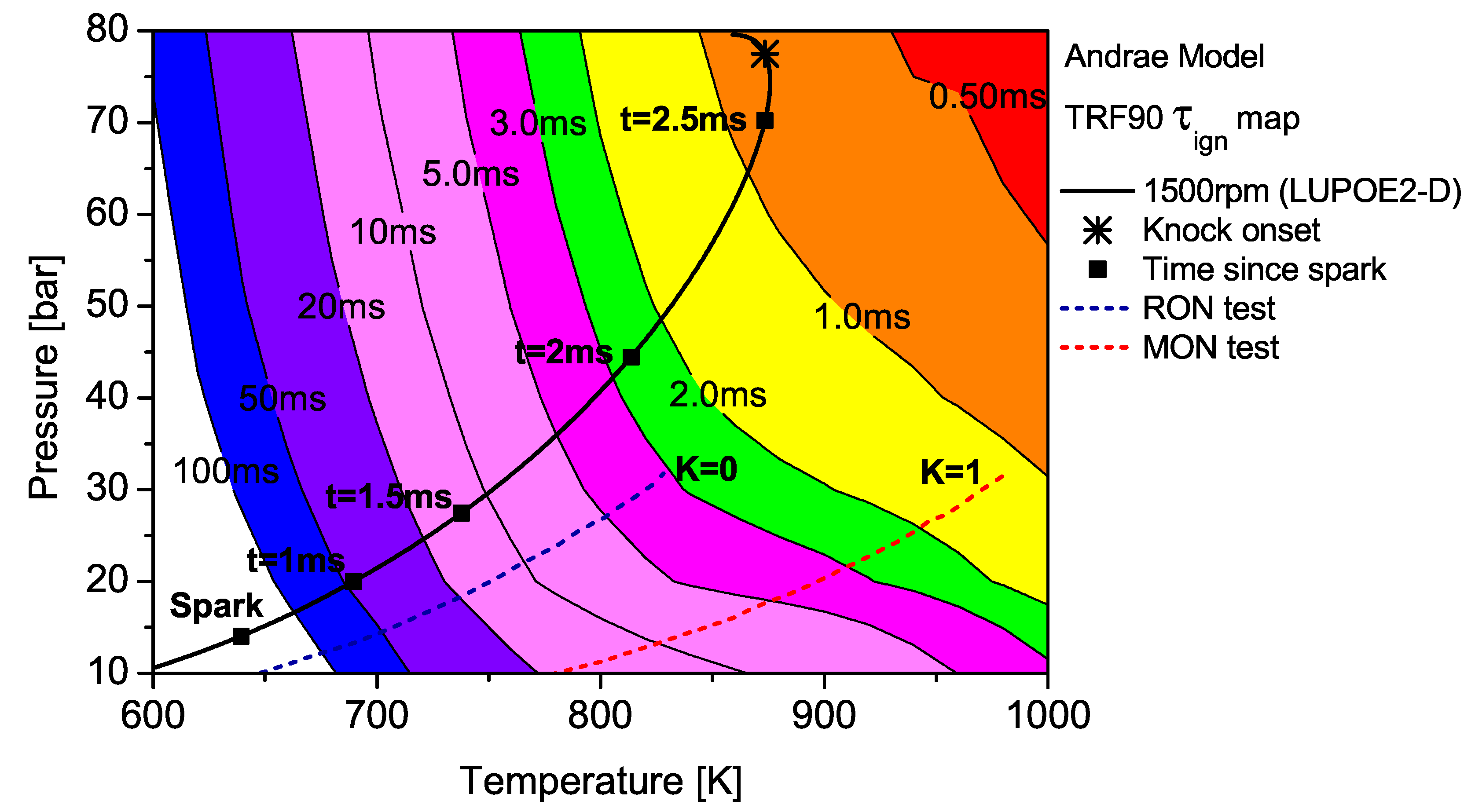
The contour map of TRF90 as predicted by the Andrae model superimposed by the unburned zone history of LUPOE2-D and typical RON/MON tests.
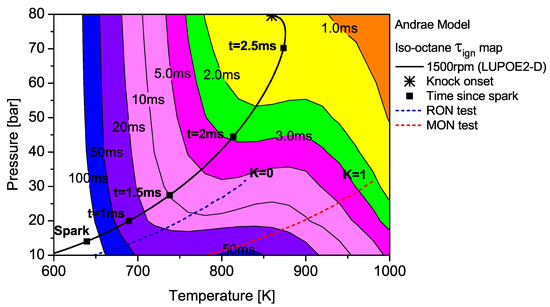
Figure 7.
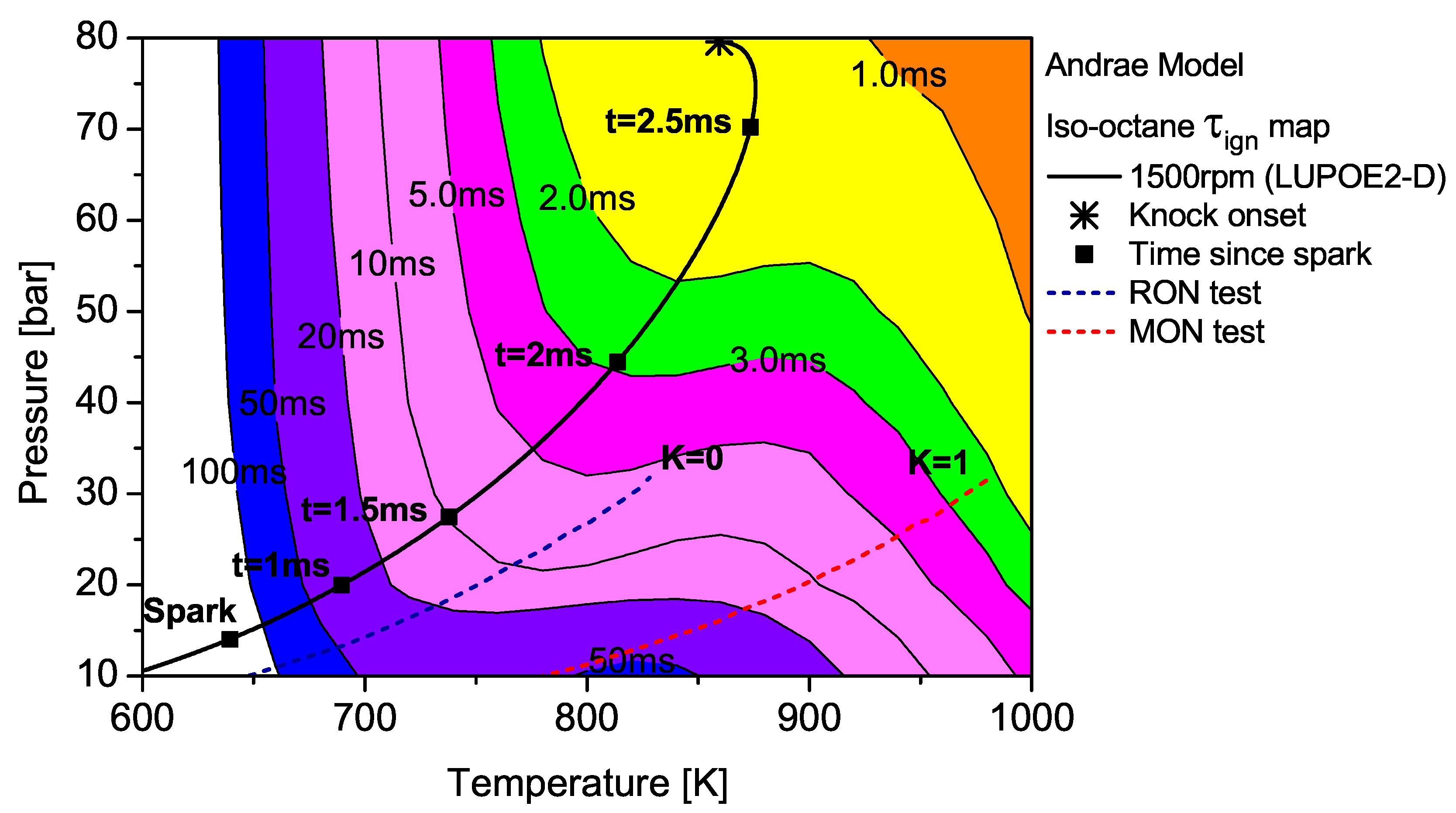
The contour map of iso-octane as predicted by the Andrae model superimposed by the unburned zone history of LUPOE2-D and typical RON/MON tests.
The end gas thermodynamic path in LUPOE2-D as well as the RON and MON tests are superimposed on the maps for PRF90 and TRF90; the latter calculated using Andrae’s model. The differences in fuel reactivity are shown by the shape of the contours and the extent to which the regions of different span. In the case of PRF90, the LUPOE2-D trajectory follows a path before the start of NTC phase where the regions of shorter are met earlier. Combustion phasing with respect to the top dead centre (TDC) is such that the regions of longer are passed relatively slowly as the pressure increase is initially slower. This phase of combustion corresponds to the period of the initial acceleration of the flame after spark until it reaches a steady turbulent burning velocity after which it starts to decelerate approaching the wall [26].
The integration of the individual along an engine’s trajectory using the Livengood-Wu integral suggests that it is the short ignition delay region towards the later stages of combustion when the are at their highest, which contribute the most in producing ignition. Comparison of the contours for PRF90, TRF90 and iso-octane shows that the regions of short are situated at lower domain for PRF90 and a significant portion of the LUPOE2-D trajectory lies in these critical regions bringing the knock onset to an earliest value of 2.8 CA after TDC (aTDC) among the four fuels studied. Iso-octane shows the latest knock onset as the trajectory stays within fairly long regions, Figure 7.
The end gas thermodynamic path in a RON test passes the same amount of time in regions of very similar for both PRF90 and TRF90, thus giving them the same RON values. This means that PRF90 can be regarded as a surrogate for TRF90 and ULG90 at only such the unburned zone conditions as encountered in the RON test. Similarly a PRF with octane number of 84.7 may be regarded as a surrogate for ULG90 at conditions of MON test. However, a single multi-component blend can be used to reproduce the autoignition behaviour of a gasoline at both RON and MON test conditions. Such a blend which has been matched to the RON/MON of a gasoline will have the same reactivity for only a narrow region of the landscape. It is therefore tempting to consider such a blend as a surrogate at all conditions. The fact that most modern SI engines operate with temperature and pressure history very similar to LUPOE2-D or at even higher pressures resulting from turbocharging, RON and MON are not sufficient to characterise auto-ignition of gasoline in such engines. Moreover, weaknesses in the empirical octane number models and the lack of ignition delay time data for gasolines reduces the surrogate formulation for SI engine autoignition to guess work.
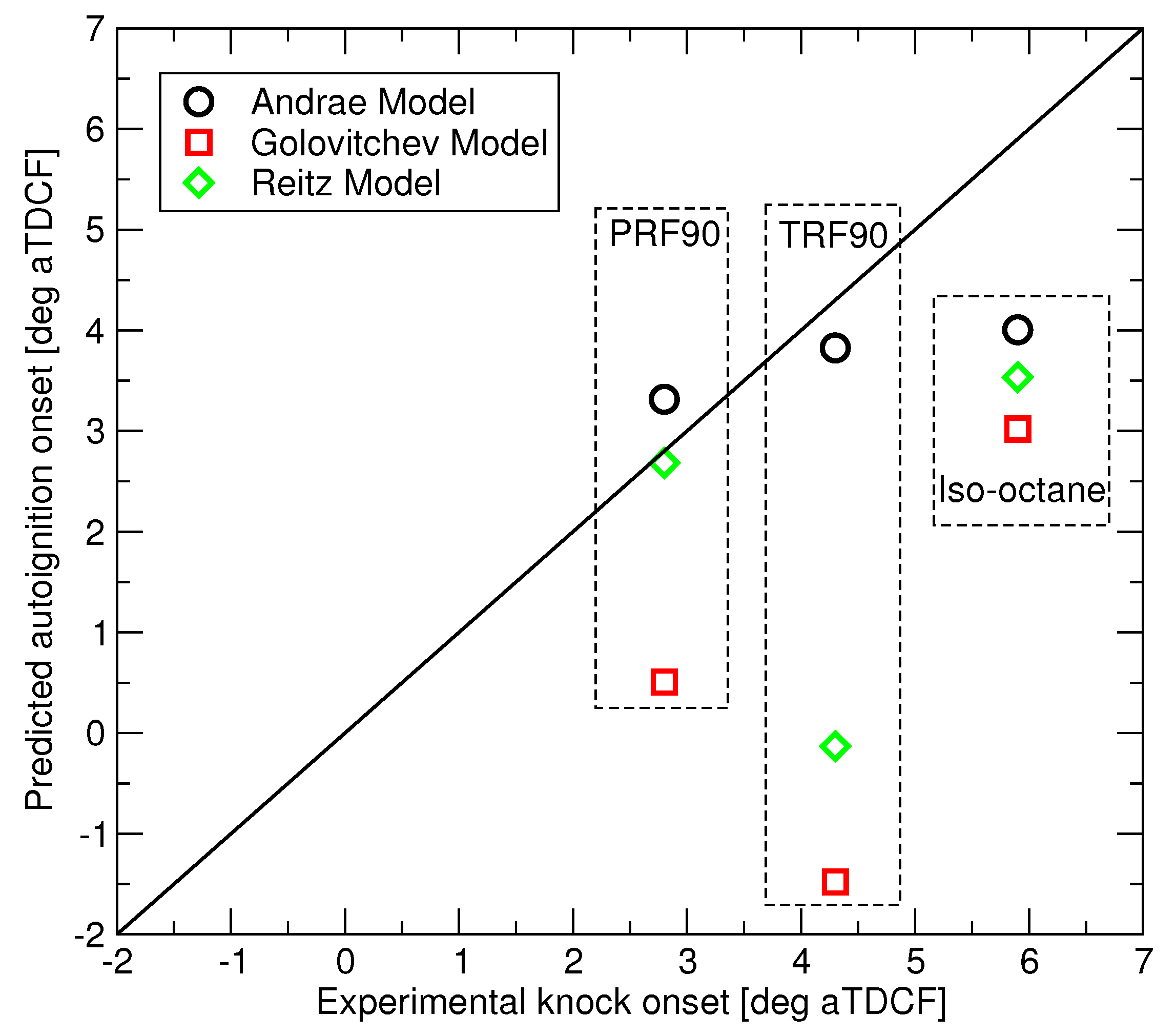
Autoignition predictions of the three RON 90 fuels have been made for the LUPOE2-D conditions of Figure 3 using the three reduced mechanisms and the comparison to the observed knock onsets is presented in Figure 8. The three fuels are subject to that same end gas and equivalence ratio history. Since LUPOE2-D was operated in skip firing mode, all trapped residual gases were expunged, hence an ideal scavenging was assumed in the modelling. Owing to this, the differences in the predicted autoignition times are caused solely by differences in the autoignition chemistry of the three fuels. Across the range of studied conditions, the Andrae’s model appears to perform consistently better than competitors, however more accurate PRF submodel in the Reitz model produces accurate autoignition prediction in the case of PRF90. Golovitchev’s model predicts shorter delays in contrast to what can be seen in the ignition delay time calculations at the constant pressure, see Figure 1.
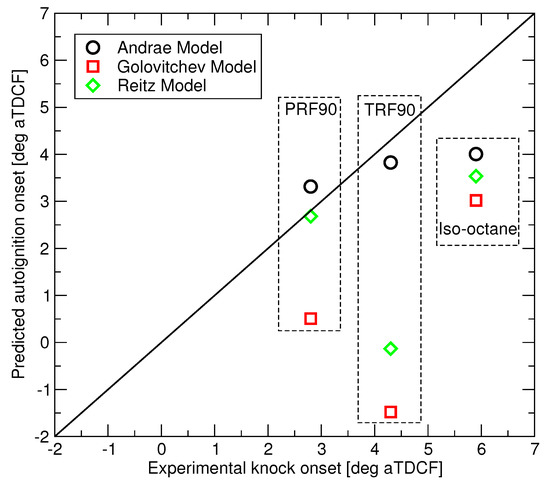
Figure 8.
Predicted vs. measured autoignition onset for the three surrogate fuels.
Table 2 lists the possible surrogates for ULG90. The constraints discussed in Section 4 have been optimised for ULG90 to formulate a properties-based surrogate. Only the Reitz model has pathways for a naphthene, here cyclohexane, a suitable surrogate component. A composition-based surrogate is also studied whose composition is the same as that of the major gasoline constituent families. A tri-component TRF with the same RON and MON as ULG90 has also been studied. The autoignition onsets of the composition and properties based surrogates for the ULG90 history in LUPOE2-D have been simulated using the Reitz model. The autoignition onset of the TRF has been predicted by the three mechanisms. It is found that it is the TRF with Andrae model which predicts the closest autoignition to that of the ULG90. Among the two 4-component surrogates, it is the composition based surrogate which yields predictions closer to the observed knock onset.
Table 2.
Key properties and volumetric composition of ULG90 and its surrogates. Predicted surrogate autoignition onsets for the ULG90 condition of Figure 3 have also been tabulated.
7. Conclusions
It has been demonstrated that reduced chemical kinetics can feasibly be used in the prediction of gasoline autoignition in a SI engine by approximating its properties by a surrogate. The choice of the mechanism and the surrogate affect greatly the prediction accuracy. The choice of the surrogate components is however limited by the availability of chemical kinetics schemes for their oxidation. Although the prediction of octane numbers is important in the formulation of a surrogate but matching of the RON and MON does not guarantee the correct reproduction of gasoline ignition behaviour in the surrogate as the modern SI engines operate at conditions much different form the RON and MON tests. Moreover, a compositional fidelity of the surrogate to that of the gasoline is also important in producing the correct autoignition behaviour in the surrogate.
The mechanisms generally over-predict the ignition delay time at low temperatures and pressure of about 40 bar. However, earlier autoignition onsets in an engine have been predicted by the three models; in particular the Golovitchev and Reitz models, indicating shorter ignition delay time predictions at lower pressures. There is still a need of validating reduced mechanisms at conditions which are found in modern downsized, turbocharged SI engines.
Author Contributions
Conceptualization and methodology: A.A.B.; software: A.F.K. and A.A.B.; validation, formal analysis, and investigation: A.F.K. and P.J.R.; data curation: A.A.B.; writing—original draft preparation A.F.K.; writing—review and editing: A.A.B.; visualization A.F.K.; supervision: A.A.B.
Acknowledgments
Authors gratefully acknowledge the financial support from EPSRC (Engineering and Physical Sciences Research Council), and Mahle Powertrain UK Ltd. Northampton UK, which funded a part of this work. Discussions with Jens Neumeister and Dave OudeNijeweme on the knocking tendency of modern high performance SI engines were useful. Many stimulating discussions with C.G.W. Sheppard, his inspiration and guidance during the experimental work were invaluable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Khan, A.F.; Burluka, A.; Neumeister, J.; OudeNijeweme, D.; Freeland, P.; Mitcalf, J. Combustion and Autoignition Modelling in a Turbocharged SI Engine. SAE Int. J. Engines 2016, 9, 2079–2090. [Google Scholar] [CrossRef]
- Bozza, F.; Siano, D.; Torella, E. Cycle-by-Cycle Analysis, Knock Modeling and Spark-Advance Setting of a “Downsized” Spark-Ignition Turbocharged Engine. SAE Int. J. Engines 2009, 2, 381–389. [Google Scholar] [CrossRef]
- Mehl, M.; Faravelli, T.; Ranzi, E.; Giavazzi, F.; Scorletti, P.; Terna, D.; D’Errico, G.; Lucchini, T.; Onorati, A. Kinetic Modelling Study of Octane Number and Sensitivity of Hydrocarbon Mixtures in CFR Engines; SAE Technical Paper 2005-24-077; Consiglio Nazionale delle Ricerche: Rome, Italy, 2005. [Google Scholar] [CrossRef]
- Andrae, J.; Head, R. HCCI experiments with gasoline surrogate fuels modeled by a semidetailed chemical kinetic model. Combust. Flame 2009, 156, 842–851. [Google Scholar] [CrossRef]
- Huang, C.; Golovitchev, V.; Lipatnikov, A. Chemical Model of Gasoline-Ethanol Blends for Internal Combustion Engine Applications. In Proceedings of the SAE 2010 World Congress & Exhibition, Detroit, MI, USA, 13–15 April 2010; SAE Technical Paper 2010-01-0543. 2010. [Google Scholar]
- Ra, Y.; Reitz, R.D. A combustion model for IC engine combustion simulations with multi-component fuels. Combust. Flame 2011, 158, 69–90. [Google Scholar] [CrossRef]
- Knop, V.; Pera, C.; Duffour, F. Validation of a ternary gasoline surrogate in a CAI engine. Combust. Flame 2013, 160, 2067–2082. [Google Scholar] [CrossRef]
- Vanhove, G.; Petit, G.; Minetti, R. Experimental study of the kinetic interactions in the low-temperature autoignition of hydrocarbon binary mixtures and a surrogate fuel. Combust. Flame 2006, 145, 521–532. [Google Scholar] [CrossRef]
- Pera, C.; Knop, V. Methodology to define gasoline surrogates dedicated to auto-ignition in engines. Fuel 2012, 96, 59–69. [Google Scholar] [CrossRef]
- Gauthier, B.; Davidson, D.; Hanson, R. Shock tube determination of ignition delay times in full-blend and surrogate fuel mixtures. Combust. Flame 2004, 139, 300–311. [Google Scholar] [CrossRef]
- Knop, V.; Loos, M.; Pera, C.; Jeuland, N. A linear-by-mole blending rule for octane numbers of n-heptane/iso-octane/toluene mixtures. Fuel 2014, 115, 666–673. [Google Scholar] [CrossRef]
- Mehl, M.; Chen, J.Y.; Pitz, W.J.; Sarathy, S.M.; Westbrook, C.K. An Approach for Formulating Surrogates for Gasoline with Application toward a Reduced Surrogate Mechanism for CFD Engine Modeling. Energy Fuels 2011, 25, 5215–5223. [Google Scholar] [CrossRef]
- Kukkadapu, G.; Kumar, K.; Sung, C.J.; Mehl, M.; Pitz, W.J. Autoignition of gasoline and its surrogates in a rapid compression machine. Proc. Combust. Inst. 2013, 34, 345–352. [Google Scholar] [CrossRef]
- Fikri, M.; Herzler, J.; Starke, R.; Schulz, C.; Roth, P.; Kalghatgi, G. Autoignition of gasoline surrogates mixtures at intermediate temperatures and high pressures. Combust. Flame 2008, 152, 276–281. [Google Scholar] [CrossRef]
- Cancino, L.; Fikri, M.; Oliveira, A.; Schulz, C. Autoignition of gasoline surrogate mixtures at intermediate temperatures and high pressures: Experimental and numerical approaches. Proc. Combust. Inst. 2009, 32, 501–508. [Google Scholar] [CrossRef]
- Khan, A.F.; Burluka, A.A. An investigation of various chemical kinetic models for the prediction of autoignition in HCCI engine. In Proceedings of the ASME 2012 Internal Combustion Engine Division Fall Technical Conference, Vancouver, BC, Canada, 23–26 September 2012; pp. 737–745. [Google Scholar]
- Fieweger, K.; Blumenthal, R.; Adomeit, G. Self-ignition of S.I. engine model fuels: A shock tube investigation at high pressure. Combust. Flame 1997, 109, 599–619. [Google Scholar] [CrossRef]
- Hartmann, M.; Gushterova, I.; Fikri, M.; Schulz, C.; Schießl, R.; Maas, U. Auto-ignition of toluene-doped n-heptane and iso-octane/air mixtures: High-pressure shock-tube experiments and kinetics modeling. Combust. Flame 2011, 158, 172–178. [Google Scholar] [CrossRef]
- Davidson, D.; Gauthier, B.; Hanson, R. Shock tube ignition measurements of iso-octane/air and toluene/air at high pressures. Proc. Combust. Inst. 2005, 30, 1175–1182. [Google Scholar] [CrossRef]
- Cash, J.R. Review Paper: Efficient numerical methods for the solution of stiff initial-value problems and differential algebraic equations. Proc. R. Soc. Lond. Ser. A Math. Phys. Eng. Sci. 2003, 459, 797–815. [Google Scholar] [CrossRef]
- Burluka, A. Combustion in a Spark Ignition Engine. In Handbook of Combustion, Volueme 3: Gaseous and Liquid Fuels; Wiley-VCH Verlag: Weinheim, Germany, 2010; Chapter 13; pp. 389–414. [Google Scholar]
- Ghosh, P.; Hickey, K.; Jaffe, S. Development of a detailed gasoline composition-based octane model. Ind. Eng. Chem. Res. 2006, 45, 337–345. [Google Scholar] [CrossRef]
- Foong, T.M.; Morganti, K.J.; Brear, M.J.; da Silva, G.; Yang, Y.; Dryer, F.L. The octane numbers of ethanol blended with gasoline and its surrogates. Fuel 2014, 115, 727–739. [Google Scholar] [CrossRef]
- Morgan, N.; Smallbone, A.; Bhave, A.; Kraft, M.; Cracknell, R.; Kalghatgi, G. Mapping surrogate gasoline compositions into RON/MON space. Combust. Flame 2010, 157, 1122–1131. [Google Scholar] [CrossRef]
- Roberts, P.; Sheppard, C. The Influence of Residual Gas NO Content on Knock Onset of Iso-Octane, PRF, TRF and ULG Mixtures in SI Engines. Sae Int. J. Engines 2013, 6, 2028–2043. [Google Scholar] [CrossRef]
- Liu, K.; Burluka, A.; Sheppard, C. Turbulent flame and mass burning rate in a spark ignition engine. Fuel 2013, 107, 202–208. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).