
Expression of Transposable Elements throughout the Fasciola hepatica Trematode Life Cycle

 , ,
, ,
Abstract
:1. Introduction
2. Results
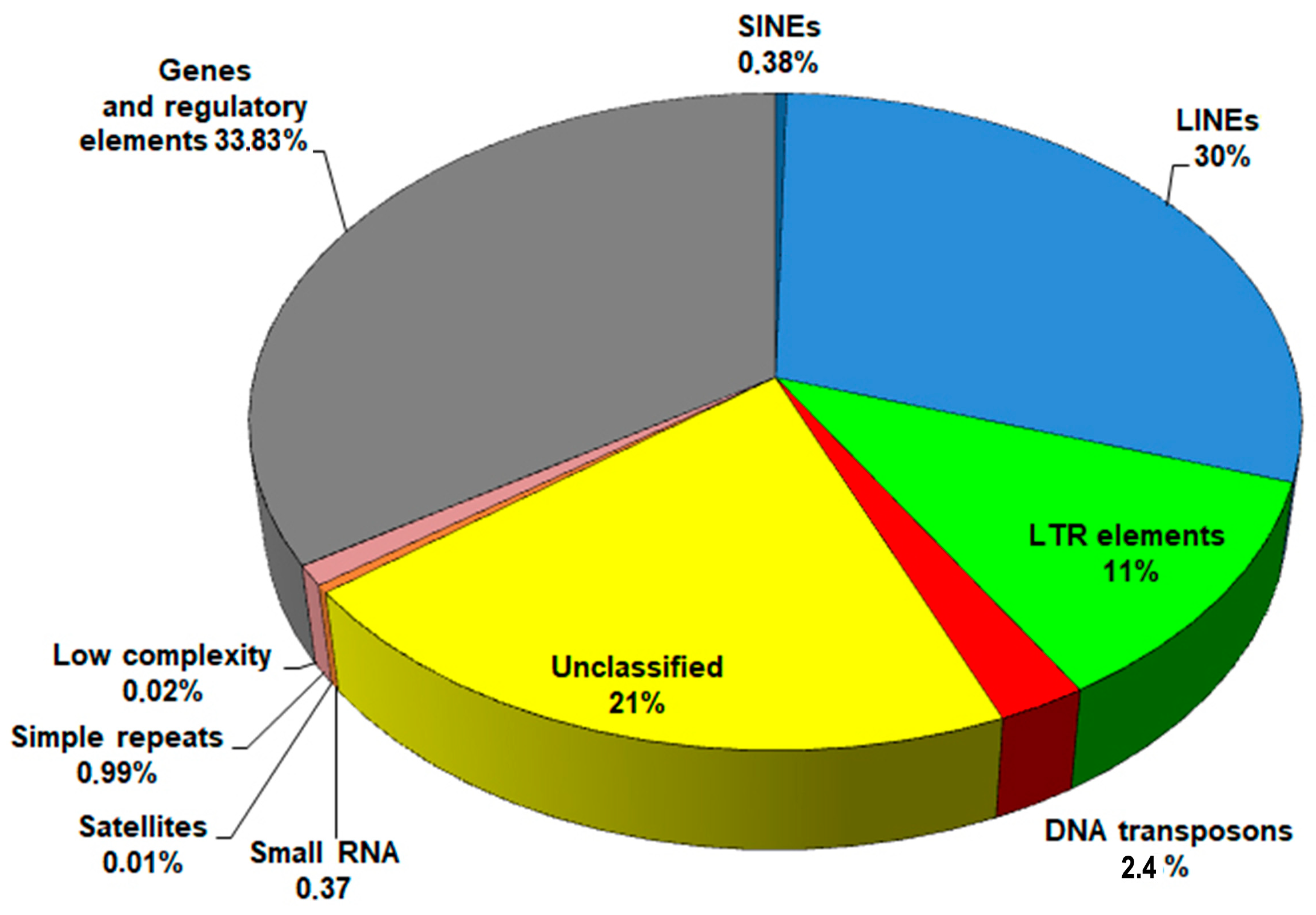
2.1. Repeat Content in the F. hepatica Genome
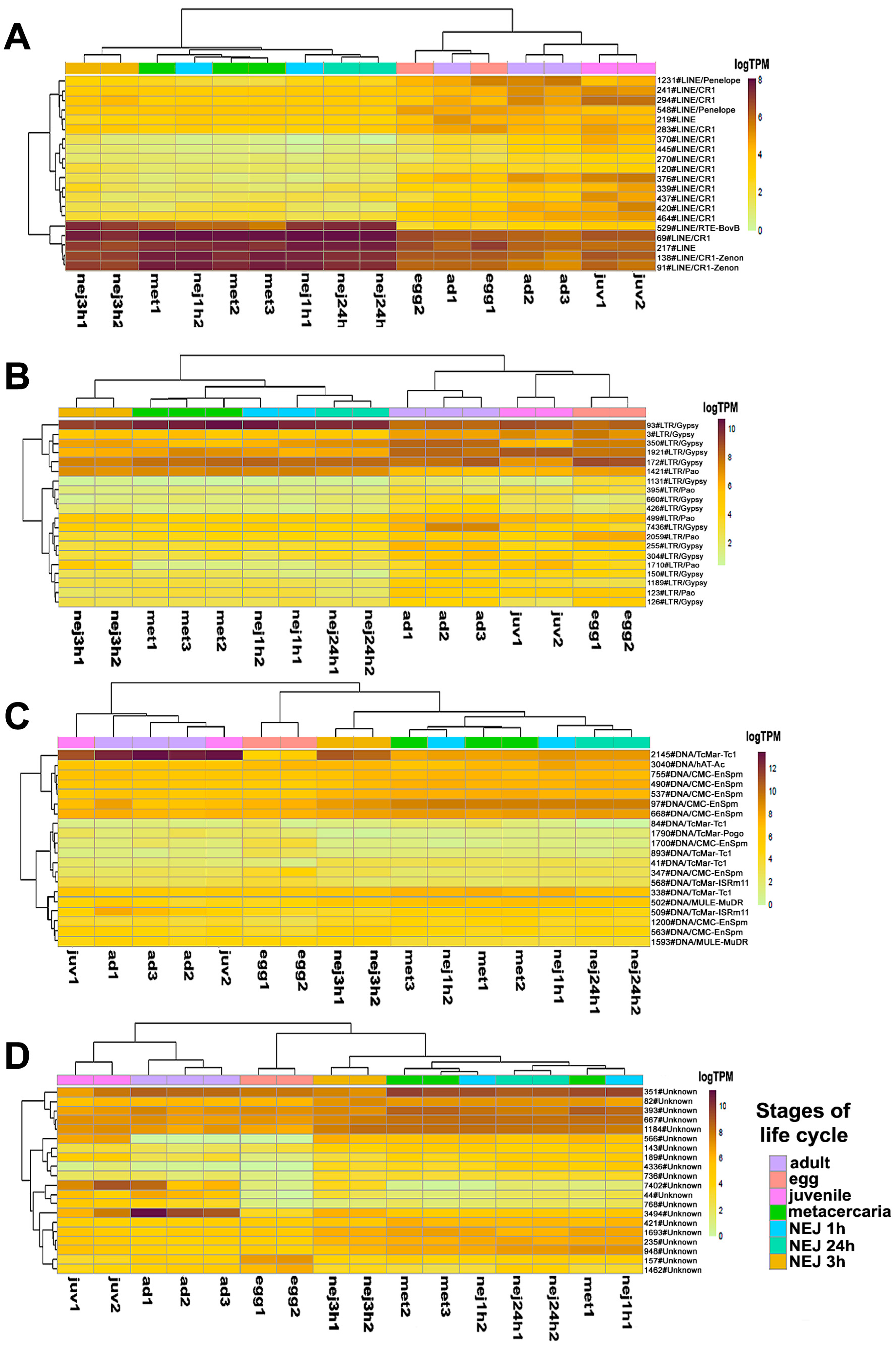
2.2. Transcription of Repetitive Elements and Candidate TEs with Stage-Specific Expression
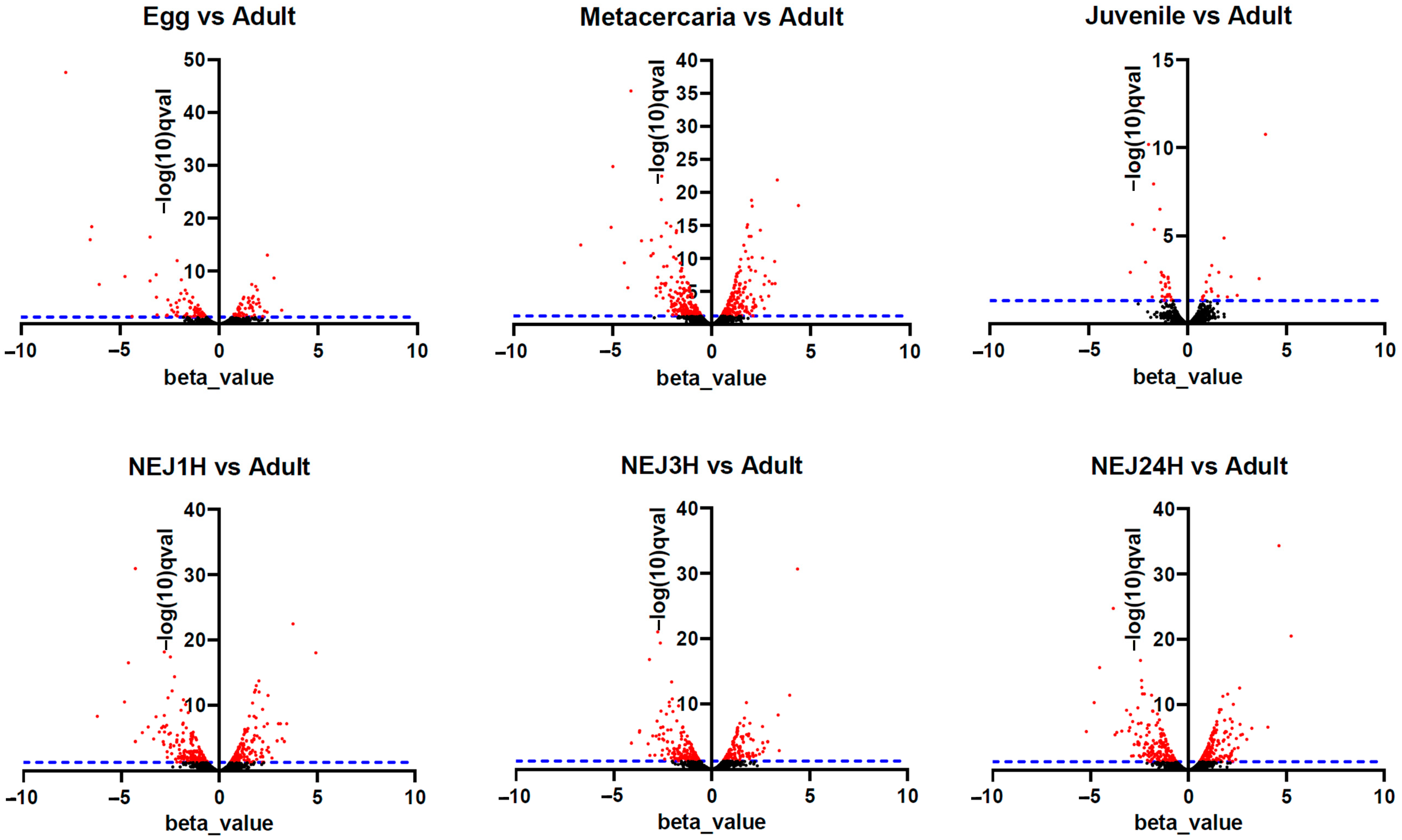
2.3. Differential Expression Analysis of Transposable Elements
3. Discussion
4. Materials and Methods
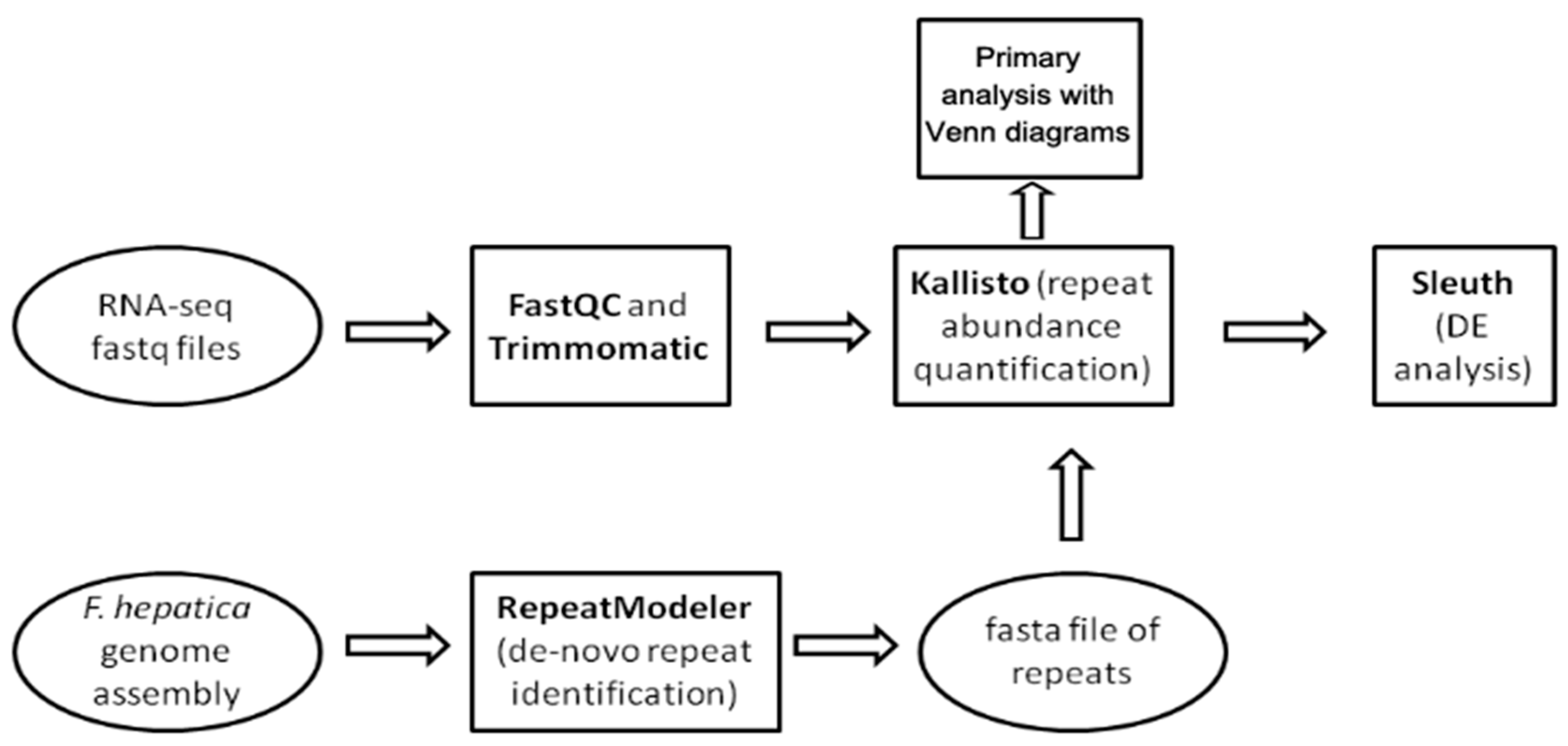
4.1. Genomic Data and Repetitive Element Discovery
4.2. RNA-Seq Data Preprocessing
4.3. Differential Expression Analysis of Transposable Elements
4.4. Identification of Transcription Factor Binding Sites in Stage-Specific Upregulated TEs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tidman, R.; Kanankege, K.S.T.; Bangert, M.; Abela-Ridder, B. Global prevalence of 4 neglected foodborne trematodes targeted for control by WHO: A scoping review to highlight the gaps. PLoS Negl. Trop. Dis. 2023, 17, e0011073. [Google Scholar] [CrossRef]
- Brindley, P.J.; da Costa, J.M.C.; Sripa, B. Why does infection with some helminths cause cancer? Trends Cancer 2015, 1, 174–182. [Google Scholar] [CrossRef]
- Efared, B.; Bako, A.B.A.; Idrissa, B.; Alhousseini, D.; Boureima, H.S.; Sodé, H.C.; Nouhou, H. Urinary bladder Schistosoma haematobium-related squamous cell carcinoma: A report of two fatal cases and literature review. Trop. Dis. Travel Med. Vaccines 2022, 8, 3. [Google Scholar] [CrossRef]
- von Bülow, V.; Lichtenberger, J.; Grevelding, C.G.; Falcone, F.H.; Roeb, E.; Roderfeld, M. Does Schistosoma Mansoni Facilitate Carcinogenesis? Cells 2021, 10, 1982. [Google Scholar] [CrossRef]
- Sripa, B.; Brindley, P.J.; Mulvenna, J.; Laha, T.; Smout, M.J.; Mairiang, E.; Bethony, J.M.; Loukas, A. The tumorigenic liver fluke Opisthorchis viverrini—Multiple pathways to cancer. Trends Parasitol. 2012, 28, 395–407. [Google Scholar] [CrossRef]
- Kim, T.-S.; Pak, J.H.; Kim, J.-B.; Bahk, Y.Y. Clonorchis sinensis, an oriental liver fluke, as a human biological agent of cholangiocarcinoma: A brief review. BMB Rep. 2016, 49, 590–597. [Google Scholar] [CrossRef]
- Aksoy, D.Y.; Kerimoglu, U.; Oto, A.; Erguven, S.; Arslan, S.; Unal, S.; Batman, F.; Bayraktar, Y. Infection with Fasciola hepatica. Clin. Microbiol. Infect. 2005, 11, 859–861. [Google Scholar] [CrossRef]
- Nesterenko, M.A.; Starunov, V.V.; Shchenkov, S.V.; Maslova, A.R.; Denisova, S.A.; Granovich, A.I.; Dobrovolskij, A.A.; Khalturin, K.V. Molecular signatures of the rediae, cercariae and adult stages in the complex life cycles of parasitic flatworms (Digenea: Psilostomatidae). Parasites Vectors 2020, 13, 559. [Google Scholar] [CrossRef]
- Brindley, P.J. The molecular biology of schistosomes. Trends Parasitol. 2005, 21, 533–536. [Google Scholar] [CrossRef]
- Gobert, G.N.; Moertel, L.; Brindley, P.J.; McManus, D.P. Developmental gene expression profiles of the human pathogen Schistosoma japonicum. BMC Genom. 2009, 10, 128. [Google Scholar] [CrossRef]
- Zhang, X.X.; Cong, W.; Elsheikha, H.M.; Liu, G.H.; Ma, J.G.; Huang, W.Y.; Zhao, Q.; Zhu, X.Q. De novo transcriptome sequencing and analysis of the juvenile and adult stages of Fasciola gigantica. Infect. Genet. Evol. 2017, 51, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Cancela, M.; Ruétalo, N.; Dell’Oca, N.; da Silva, E.; Smircich, P.; Rinaldi, G.; Roche, L.; Carmona, C.; Alvarez-Valín, F.; Zaha, A.; et al. Survey of transcripts expressed by the invasive juvenile stage of the liver fluke Fasciola hepatica. BMC Genom. 2010, 11, 227. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, E.J.R.; Dasilva, L.F.; Pires, D.S.; Lavezzo, G.M.; Pereira, A.S.A.; Amaral, M.S.; Verjovski-Almeida, S. The Schistosoma mansoni genome encodes thousands of long non-coding RNAs predicted to be functional at different parasite life-cycle stages. Sci. Rep. 2017, 7, 10508. [Google Scholar] [CrossRef]
- Vasconcelos, E.J.R.; Mesel, V.C.; DaSilva, L.F.; Pires, D.S.; Lavezzo, G.M.; Pereira, A.S.A.; Amaral, M.S.; Verjovski-Almeida, S. Atlas of Schistosoma mansoni long non-coding RNAs and their expression correlation to protein-coding genes. Database 2018, 2018, bay068. [Google Scholar] [CrossRef] [PubMed]
- McVeigh, P.; McCammick, E.; Robb, E.; Brophy, P.; Morphew, R.M.; Marks, N.J.; Maule, A.G. Discovery of long non-coding RNAs in the liver fluke, Fasciola hepatica. PLoS Negl. Trop. Dis. 2023, 17, e0011663. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Kronenberg, Z.; Lynch, V.J.; Zhuo, X.; Ramsay, L.A.; Bourque, G.; Yandell, M.; Feschotte, C. Transposable Elements Are Major Contributors to the Origin, Diversification, and Regulation of Vertebrate Long Noncoding RNAs. PLoS Genet. 2013, 9, e1003470. [Google Scholar] [CrossRef] [PubMed]
- Fort, V.; Khelifi, G.; Hussein, S.M.I. Long non-coding RNAs and transposable elements: A functional relationship. Biochim. Biophys. Acta-Mol. Cell Res. 2021, 1868, 118837. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Warren, I.A.; Naville, M.; Chalopin, D.; Levin, P.; Berger, C.S.; Galiana, D.; Volff, J.N. Evolutionary impact of transposable elements on genomic diversity and lineage-specific innovation in vertebrates. Chromosom. Res. 2015, 23, 505–531. [Google Scholar] [CrossRef]
- Chalopin, D.; Naville, M.; Plard, F.; Galiana, D.; Volff, J.N. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol. Evol. 2015, 7, 567–580. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Huang, C.R.L.; Burns, K.H.; Boeke, J.D. Active transposition in genomes. Annu. Rev. Genet. 2012, 46, 651–675. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2016, 18, 71–86. [Google Scholar] [CrossRef]
- Kazazian, H.H. Mobile elements: Drivers of genome evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef]
- Deininger, P.L.; Moran, J.V.; Batzer, M.A.; Kazazian, H.H. Mobile elements and mammalian genome evolution. Curr. Opin. Genet. Dev. 2003, 13, 651–658. [Google Scholar] [CrossRef]
- Chénais, B.; Caruso, A.; Hiard, S.; Casse, N. The impact of transposable elements on eukaryotic genomes: From genome size increase to genetic adaptation to stressful environments. Gene 2012, 509, 7–15. [Google Scholar] [CrossRef]
- Cwiklinski, K.; Dalton, J.P.; Dufresne, P.J.; La Course, J.; Williams, D.J.; Hodgkinson, J.; Paterson, S. The Fasciola hepatica genome: Gene duplication and polymorphism reveals adaptation to the host environment and the capacity for rapid evolution. Genome Biol. 2015, 16, 71. [Google Scholar] [CrossRef]
- Mas-Coma, S.; Valero, M.A.; Bargues, M.D. Fascioliasis. Adv. Exp. Med. Biol. 2019, 1154, 71–103. [Google Scholar] [CrossRef]
- McNulty, S.N.; Tort, J.F.; Rinaldi, G.; Fischer, K.; Rosa, B.A.; Smircich, P.; Fontenla, S.; Choi, Y.J.; Tyagi, R.; Hallsworth-Pepin, K.; et al. Genomes of Fasciola hepatica from the Americas Reveal Colonization with Neorickettsia Endobacteria Related to the Agents of Potomac Horse and Human Sennetsu Fevers. PLoS Genet. 2017, 13, e1006537. [Google Scholar] [CrossRef]
- Fontenla, S.; Langleib, M.; de la Torre-Escudero, E.; Domínguez, M.F.; Robinson, M.W.; Tort, J. Role of Fasciola hepatica Small RNAs in the Interaction with the Mammalian Host. Front. Cell. Infect. Microbiol. 2022, 11, 812141. [Google Scholar] [CrossRef]
- Herron, C.M.; O’Connor, A.; Robb, E.; McCammick, E.; Hill, C.; Marks, N.J.; Robinson, M.W.; Maule, A.G.; McVeigh, P. Developmental Regulation and Functional Prediction of microRNAs in an Expanded Fasciola hepatica miRNome. Front. Cell. Infect. Microbiol. 2022, 12, 811123. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef]
- Goubert, C.; Zevallos, N.A.; Feschotte, C. Contribution of unfixed transposable element insertions to human regulatory variation. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190331. [Google Scholar] [CrossRef]
- Barth, N.K.H.; Li, L.; Taher, L. Independent Transposon Exaptation Is a Widespread Mechanism of Redundant Enhancer Evolution in the Mammalian Genome. Genome Biol. Evol. 2020, 12, 1–17. [Google Scholar] [CrossRef]
- Nicolau, M.; Picault, N.; Moissiard, G. The Evolutionary Volte-Face of Transposable Elements: From Harmful Jumping Genes to Major Drivers of Genetic Innovation. Cells 2021, 10, 2952. [Google Scholar] [CrossRef]
- Silveira, G.O.; Coelho, H.S.; Pereira, A.S.A.; Miyasato, P.A.; Santos, D.W.; Maciel, L.F.; Olberg, G.G.G.; Tahira, A.C.; Nakano, E.; Oliveira, M.L.S.; et al. Long non-coding RNAs are essential for Schistosoma mansoni pairing-dependent adult worm homeostasis and fertility. PLoS Pathog. 2023, 19, e1011369. [Google Scholar] [CrossRef]
- Wang, S.S.; Chen, D.; He, J.J.; Zheng, W.B.; Tian, A.L.; Zhao, G.H.; Elsheikha, H.M.; Zhu, X.Q. Fasciola gigantica–Derived Excretory-Secretory Products Alter the Expression of mRNAs, miRNAs, lncRNAs, and circRNAs Involved in the Immune Response and Metabolism in Goat Peripheral Blood Mononuclear Cells. Front. Immunol. 2021, 12, 653755. [Google Scholar] [CrossRef]
- Fontenla, S.; Rinaldi, G.; Smircich, P.; Tort, J.F. Conservation and diversification of small RNA pathways within flatworms. BMC Evol. Biol. 2017, 17, 215. [Google Scholar] [CrossRef]
- Jardim Poli, P.; Fischer-Carvalho, A.; Tahira, A.C.; Chan, J.D.; Verjovski-Almeida, S.; Sena Amaral, M. Long Non-Coding RNA Levels Are Modulated in Schistosoma mansoni Following In Vivo Praziquantel Exposure. Non-Coding RNA 2024, 10, 27. [Google Scholar] [CrossRef]
- Laha, T.; Loukas, A.; Smyth, D.J.; Copeland, C.S.; Brindley, P.J. The fugitive LTR retrotransposon from the genome of the human blood fluke, Schistosoma mansoni. Int. J. Parasitol. 2004, 34, 1365–1375. [Google Scholar] [CrossRef]
- Copeland, C.S.; Brindley, P.J.; Heyers, O.; Michael, S.F.; Johnston, D.A.; Williams, D.L.; Ivens, A.C.; Kalinna, B.H. Boudicca, a retrovirus-like long terminal repeat retrotransposon from the genome of the human blood fluke Schistosoma mansoni. J. Virol. 2003, 77, 6153–6166. [Google Scholar] [CrossRef]
- Brindley, P.J.; Copeland, C.S.; Kalinna, B.H. Schistosome Retrotransposons BT—In Schistosomiasis. In World Class Parasites; Secor, W.E., Colley, D.G., Eds.; Springer: Boston, MA, USA, 2005; pp. 13–26. ISBN 978-0-387-23362-8. [Google Scholar]
- Galaktionov, N.K.; Solovyeva, A.I.; Fedorov, A.V.; Podgornaya, O.I. Trematode Himasthla elongata mariner element (Hemar): Structure and applications. J. Exp. Zool. Part B Mol. Dev. Evol. 2014, 322, 142–155. [Google Scholar] [CrossRef]
- Solovyeva, A.; Levakin, I.; Zorin, E.; Adonin, L.; Khotimchenko, Y.; Podgornaya, O. Transposons-based clonal diversity in trematode involves parts of cr1 (Line) in eu-and heterochromatin. Genes 2021, 12, 1129. [Google Scholar] [CrossRef]
- Korsunenko, A.; Chrisanfova, G.; Arifov, A.; Ryskov, A.; Semyenova, S. Characterization of randomly amplified polymorphic DNA (RAPD) fragments revealing clonal variability in cercariae of avian schistosome Trichobilharzia szidati (Trematoda: Schistosomatidae). Open J. Genet. 2013, 2013, 141–158. [Google Scholar] [CrossRef]
- DeMarco, R.; Kowaltowski, A.T.; Machado, A.A.; Soares, M.B.; Gargioni, C.; Kawano, T.; Rodrigues, V.; Madeira, A.M.B.N.; Wilson, R.A.; Menck, C.F.M.; et al. Erratum: Saci-1, -2, and -3 and Perere, Four Novel Retrotransposons with High Transcriptional Activities from the Human Parasite Schistosoma mansoni. J. Virol. 2004, 78, 2967–2978. [Google Scholar] [CrossRef]
- Choi, Y.J.; Fontenla, S.; Fischer, P.U.; Le, T.H.; Costábile, A.; Blair, D.; Brindley, P.J.; Tort, J.F.; Cabada, M.M.; Mitreva, M. Adaptive Radiation of the Flukes of the Family Fasciolidae Inferred from Genome-Wide Comparisons of Key Species. Mol. Biol. Evol. 2020, 37, 84–99. [Google Scholar] [CrossRef]
- Luo, X.; Cui, K.; Wang, Z.; Li, Z.; Wu, Z.; Huang, W.; Zhu, X.Q.; Ruan, J.; Zhang, W.; Liu, Q. High-quality reference genome of Fasciola gigantica: Insights into the genomic signatures of transposon-mediated evolution and specific parasitic adaption in tropical regions. PLoS Negl. Trop. Dis. 2021, 15, e0009750. [Google Scholar] [CrossRef]
- Brindley, P.J. Mobile Genetic Elements in Metazoan Parasites; CRC Press: Boca Raton, FL, USA, 2009; ISBN 9781587060939. [Google Scholar]
- Ershov, N.I.; Mordvinov, V.A.; Prokhortchouk, E.B.; Pakharukova, M.Y.; Gunbin, K.V.; Ustyantsev, K.; Genaev, M.A.; Blinov, A.G.; Mazur, A.; Boulygina, E.; et al. New insights from Opisthorchis felineus genome: Update on genomics of the epidemiologically important liver flukes. BMC Genom. 2019, 20, 399. [Google Scholar] [CrossRef]
- Berriman, M.; Haas, B.J.; LoVerde, P.T.; Wilson, R.A.; Dillon, G.P.; Cerqueira, G.C.; Mashiyama, S.T.; Al-Lazikani, B.; Andrade, L.F.; Ashton, P.D.; et al. The genome of the blood fluke Schistosoma mansoni. Nature 2009, 460, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, W.; Huang, Y.; Sun, J.; Men, J.; Liu, H.; Luo, F.; Guo, L.; Lv, X.; Deng, C.; et al. The draft genome of the carcinogenic human liver fluke Clonorchis sinensis. Genome Biol. 2011, 12, R107. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, H.; Chen, Y.; Zhang, L.; Wang, K.; Guo, J.; Huang, Z.; Zhang, B.; Huang, W.; Jin, K.; et al. The Schistosoma japonicum genome reveals features of host-parasite interplay. Nature 2009, 460, 345–351. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and its Applications. Genomics. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Jurka, J. Repbase update: A database and an electronic journal of repetitive elements. Trends Genet. 2000, 16, 418–420. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Loreto, E.L.S.; de Melo, E.S.; Wallau, G.L.; Gomes, T.M.F.F. The good, the bad and the ugly of transposable elements annotation tools. Genet. Mol. Biol. 2024, 46, e20230138. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, L.A.; Marchetto, M.C.; Caron, M.; Chen, S.H.; Busche, S.; Kwan, T.; Pastinen, T.; Gage, F.H.; Bourque, G. Conserved expression of transposon-derived non-coding transcripts in primate stem cells. BMC Genom. 2017, 18, 214. [Google Scholar] [CrossRef] [PubMed]
- Macia, A.; Blanco-Jimenez, E.; García-Pérez, J.L. Retrotransposons in pluripotent cells: Impact and new roles in cellular plasticity. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 417–426. [Google Scholar] [CrossRef]
- Fadloun, A.; Le Gras, S.; Jost, B.; Ziegler-Birling, C.; Takahashi, H.; Gorab, E.; Carninci, P.; Torres-Padilla, M.E. Chromatin signatures and retrotransposon profiling in mouse embryos reveal regulation of LINE-1 by RNA. Nat. Struct. Mol. Biol. 2013, 20, 332–338. [Google Scholar] [CrossRef]
- Chang, N.C.; Rovira, Q.; Wells, J.; Feschotte, C.; Vaquerizas, J.M. Zebrafish transposable elements show extensive diversification in age, genomic distribution, and developmental expression. Genome Res. 2022, 32, 1408–1423. [Google Scholar] [CrossRef]
- Faunes, F.; Lee-Liu, D.; Larrain, J. Expression of DNA transposable elements during nervous system development: A discussion about its possible functions. Mob. Genet. Elem. 2011, 1, 296–300. [Google Scholar] [CrossRef]
- Ansaloni, F.; Scarpato, M.; Di Schiavi, E.; Gustincich, S.; Sanges, R. Exploratory analysis of transposable elements expression in the C. elegans early embryo. BMC Bioinform. 2019, 20, 484. [Google Scholar] [CrossRef]
- Torres-Padilla, M.E. On transposons and totipotency. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190339. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, P.; Richardson, S.R.; Mager, D.L.; Faulkner, G.J. Transposable elements in the mammalian embryo: Pioneers surviving through stealth and service. Genome Biol. 2016, 17, 100. [Google Scholar] [CrossRef]
- DiRusso, J.A.; Clark, A.T. Transposable elements in early human embryo development and embryo models. Curr. Opin. Genet. Dev. 2023, 81, 102086. [Google Scholar] [CrossRef]
- Panyushev, N.; Okorokova, L.; Danilov, L.; Adonin, L. Pattern of repetitive element transcription segregate cell lineages during the embryogenesis of sea urchin strongylocentrotus purpuratus. Biomedicines 2021, 9, 1736. [Google Scholar] [CrossRef] [PubMed]
- Nesterenko, M.; Shchenkov, S.; Denisova, S.; Starunov, V. The digenean complex life cycle: Phylostratigraphy analysis of the molecular signatures. Biol. Commun. 2022, 67, 65–87. [Google Scholar] [CrossRef]
- Zhang, X.X.; Cwiklinski, K.; Hu, R.S.; Zheng, W.B.; Sheng, Z.A.; Zhang, F.K.; Elsheikha, H.M.; Dalton, J.P.; Zhu, X.Q. Complex and dynamic transcriptional changes allow the helminth Fasciola gigantica to adjust to its intermediate snail and definitive mammalian hosts. BMC Genom. 2019, 20, 729. [Google Scholar] [CrossRef]
- Pomaznoy, M.Y.; Logacheva, M.D.; Young, N.D.; Penin, A.A.; Ershov, N.I.; Katokhin, A.V.; Mordvinov, V.A. Whole transcriptome profiling of adult and infective stages of the trematode Opisthorchis felineus. Parasitol. Int. 2016, 65, 12–19. [Google Scholar] [CrossRef]
- Cho, P.Y.; Kim, T.I.; Whang, S.M.; Hong, S.J. Gene expression profile of Clonorchis sinensis metacercariae. Parasitol. Res. 2008, 102, 277–282. [Google Scholar] [CrossRef]
- Kim, H.C.; Khalil, A.M.; Jolly, E.R. LncRNAs in molluscan and mammalian stages of parasitic schistosomes are developmentally-regulated and coordinately expressed with protein-coding genes. RNA Biol. 2020, 17, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fang, F. Long Noncoding RNA Mediated Regulation in Human Embryogenesis, Pluripotency, and Reproduction. Stem Cells Int. 2022, 2022, 8051717. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Mu, H.; Wen, B.; Zhang, W.; Wei, Q.; Gao, G.; Han, J.; Cao, S. Long non-coding RNAs involved in the regulatory network during porcine pre-implantation embryonic development and iPSC induction. Sci. Rep. 2018, 8, 6649. [Google Scholar] [CrossRef] [PubMed]
- Akay, A.; Jordan, D.; Navarro, I.C.; Wrzesinski, T.; Ponting, C.P.; Miska, E.A.; Haerty, W. Identification of functional long non-coding RNAs in C. elegans. BMC Biol. 2019, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.B.; Tsitsipatis, D.; Gorospe, M. Integrated lncRNA function upon genomic and epigenomic regulation. Mol. Cell 2022, 82, 2252–2266. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, V.; Cheng, Y.; Ma, Z.; Li, D.; Xing, X.; Edge, P.; Snyder, M.P.; Wang, T. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014, 24, 1963–1976. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Feschotte, C. Volatile evolution of long noncoding RNA repertoires: Mechanisms and biological implications. Trends Genet. 2014, 30, 439–452. [Google Scholar] [CrossRef]
- Ecco, G.; Cassano, M.; Kauzlaric, A.; Duc, J.; Coluccio, A.; Offner, S.; Imbeault, M.; Rowe, H.M.; Turelli, P.; Trono, D. Transposable Elements and Their KRAB-ZFP Controllers Regulate Gene Expression in Adult Tissues. Dev. Cell 2016, 36, 611–623. [Google Scholar] [CrossRef]
- Wood, S.; Ishida, K.; Hagerty, J.R.; Karahodza, A.; Dennis, J.N.; Jolly, E.R. Characterization of Schistosome Sox Genes and Identification of a Flatworm Class of Sox Regulators. Pathogens 2023, 12, 690. [Google Scholar] [CrossRef]
- Sundaram, V.; Wysocka, J. Transposable elements as a potent source of diverse cis-regulatory sequences in mammalian genomes. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190347. [Google Scholar] [CrossRef]
- Li, Z.; Xu, H.; Li, J.; Xu, X.; Wang, J.; Wu, D.; Zhang, J.; Liu, J.; Xue, Z.; Zhan, G.; et al. Selective binding of retrotransposons by ZFP352 facilitates the timely dissolution of totipotency network. Nat. Commun. 2023, 14, 3646. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Allton, K.; Duncan, S.A.; Shen, L.; Barton, M.C. Foxa1 functions as a pioneer transcription factor at transposable elements to activate Afp during differentiation of embryonic stem cells. J. Biol. Chem. 2010, 285, 16135–16144. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Sugimoto, R.; Nakaoka, H.; Yamada, S.; Kimura, T.; Hayano, T.; Inoue, I. Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses. PLoS Genet. 2017, 13, e1006883. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinforma. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/ (accessed on 15 April 2024).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 2017, 14, 687–690. [Google Scholar] [CrossRef]
- Zhao, Y.; Stormo, G.D. Quantitative analysis demonstrates most transcription factors require only simple models of specificity. Nat. Biotechnol. 2011, 29, 480–483. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage of Life Cycle | DNA-Transposons | Retroelements | Unknown Elements |
|---|---|---|---|
| Adult | - | 809#LINE/CR1,1652#LINE/CR1, 2673#LINE/CR1 | 1132#Unk, 5212#Unk, 210#Unk, 4115#Unk, 3145#Unk, 2534#Unk, 2941#Unk, 1834#Unk, 504#Unk, 713#Unk, 392#Unk, 569#Unk, 416#Unk, 3383#Unk, 1574#Unk |
| Egg | - | 622#LINE/CR1,596#LINE/CR1, 1595#LTR/Gypsy,456#LINE/CR1 | 2633#Unk, 1040#Unk,587#Unk,515#Unk, 971#Unk, 1393#Unk, 431#Unk, 1119#Unk |
| Juvenile | - | 962#LINE/CR1, 441#LTR/Pao, 432#LINE/Penelope, 129#LINE/CR1, 313#LTR/Pao | 3067#Unk, 423#Unk,764#Unk,1693#Unk, 566#Unk,9365#Unk,3346#Unk, 2338#Unk, 280#Unk,955#Unk,4831#Unk,138#Unk,584#Unk, 196#Unk, 335#Unk,297#Unk, 246#Unk |
| Metacercaria | 487#DNA/CMC-EnSpm, 2355#DNA/TcMar-Tigger | 6300#LTR/Gypsy, 191#LINE/CR1 | 969#Unk,1704#Unk,1134#Unk,1234#Unk, 371#Unk, 867#Unk, 1263#Unk,1123#Unk, 38#Unk,1339#Unk,495#Unk,426#Unk,69#Unk |
| NEJ 24 h | - | 467#LTR/Gypsy, 183#LINE/CR1, 2677#LINE/CR1, 502#LINE/CR1 | 410#Unk, 336#Unk, 2170#Unk, 3876#Unk, 6772#Unk, 1365#Unk |
| Stage of Life Cycle | DNA-Transposons | Retroelements | Unknown Elements |
|---|---|---|---|
| Adult | - | 809#LINE/CR1 | 1132#Unk, 5212#Unk, 210#Unk, 504#Unk, 2534#Unk, 2941#Unk, 713#Unk, 392#Unk, 569#Unk, 416#Unk, 3383#Unk, 1574#Unk,1834#Unk, |
| Egg | - | 622#LINE/CR1,596#LINE/CR1, 1595#LTR/Gypsy | 2633#Unk, 1040#Unk, 587#Unk, 515#Unk, 971#Unk, 1393#Unk, 431#Unk, 1119#Unk |
| Juvenile | - | 313#LTR/Pao, 962#LINE/CR1 441#LTR/Pao, 129#LINE/CR1 | 3067#Unk, 764#Unk, 1693#Unk 566#Unk,9365#Unk,3346#Unk, 2338#Unk, 280#Unk,955#Unk 4831#Unk, 584#Unk, 196#Unk 335#Unk, 297#Unk, 246#Unk |
| Metacercaria | 2355#DNA/TcMar-Tigger | - | 969#Unk, 704#Unk, 134#Unk, 234#Unk, 371#Unk, 867#Unk, 1339#Unk, 495#Unk, 426#Unk, 69#Unk |
| Juvenile 24 h | - | 467#LTR/Gypsy, 183#LINE/CR1 2677#LINE/CR1, 502#LINE/CR1 | 410#Unk, 336#Unk, 3876#Unk 1365#Unk |
| Juvenile 1 h | - | 1677#LTR/Gypsy | 261#Unk, 4259#Unk |
| Juvenile 3 h | - | - | 1270#Unk, 892#Unk, 547#Unk |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skalon, E.K.; Panyushev, N.V.; Podgornaya, O.I.; Smolyaninova, A.R.; Solovyeva, A.I. Expression of Transposable Elements throughout the Fasciola hepatica Trematode Life Cycle. Non-Coding RNA 2024, 10, 39. https://doi.org/10.3390/ncrna10040039
Skalon EK, Panyushev NV, Podgornaya OI, Smolyaninova AR, Solovyeva AI. Expression of Transposable Elements throughout the Fasciola hepatica Trematode Life Cycle. Non-Coding RNA. 2024; 10(4):39. https://doi.org/10.3390/ncrna10040039
Chicago/Turabian StyleSkalon, Elizaveta K., Nick V. Panyushev, Olga I. Podgornaya, Anastasia R. Smolyaninova, and Anna I. Solovyeva. 2024. "Expression of Transposable Elements throughout the Fasciola hepatica Trematode Life Cycle" Non-Coding RNA 10, no. 4: 39. https://doi.org/10.3390/ncrna10040039