Evolutionary Implications of the microRNA- and piRNA Complement of Lepidodermella squamata (Gastrotricha)

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
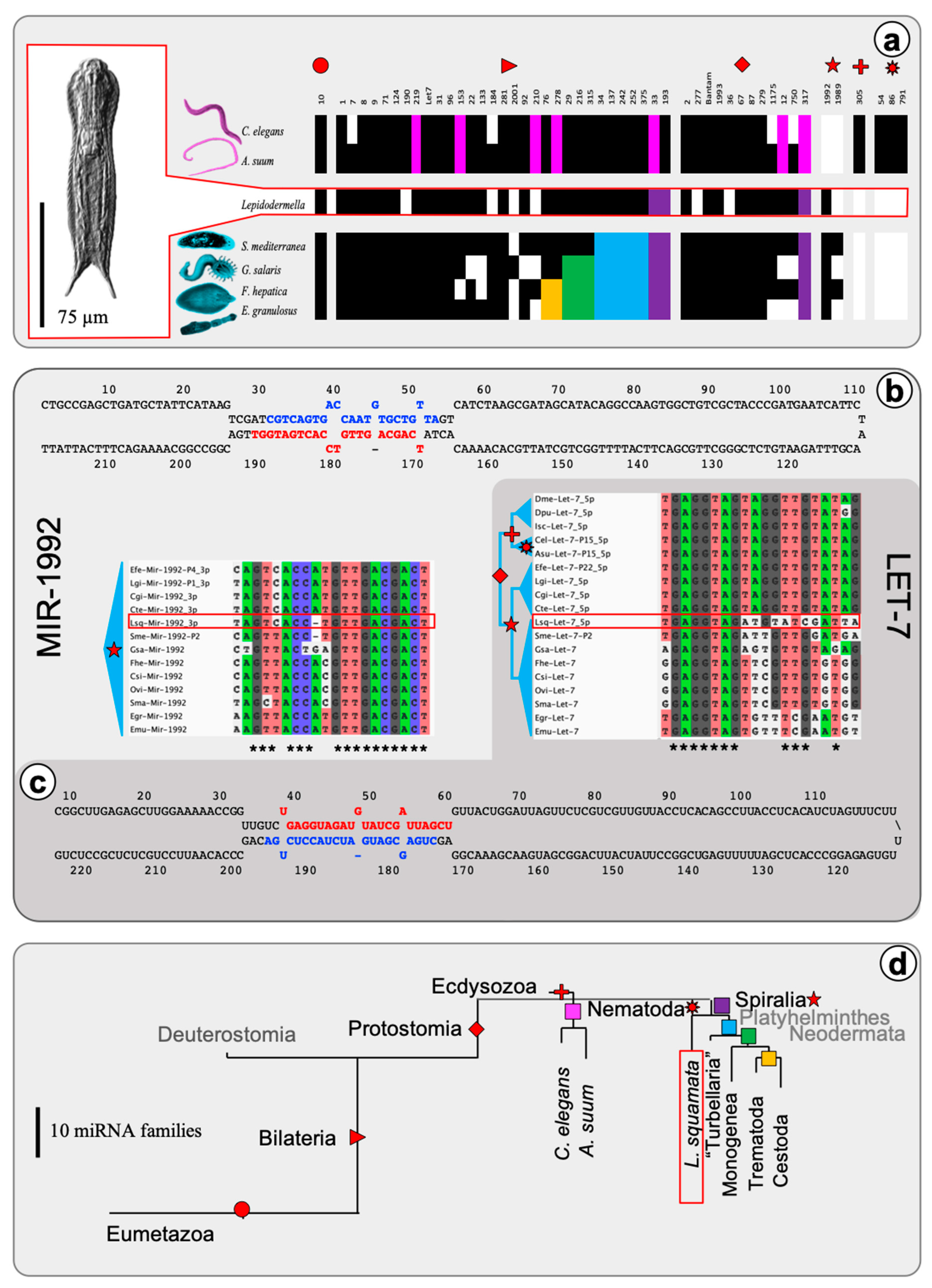
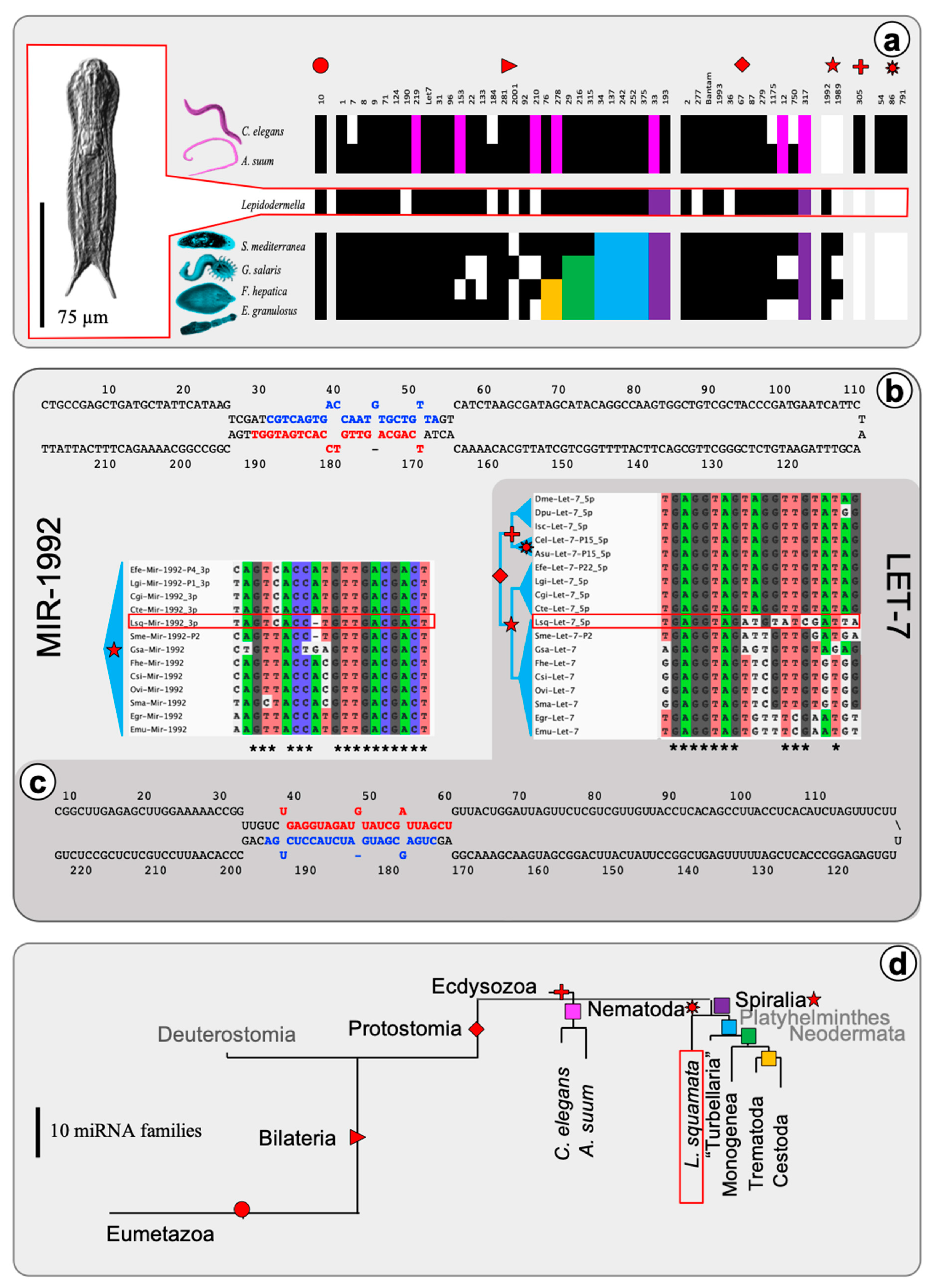
2.1. The microRNA Complement of Lepidodermella squamata
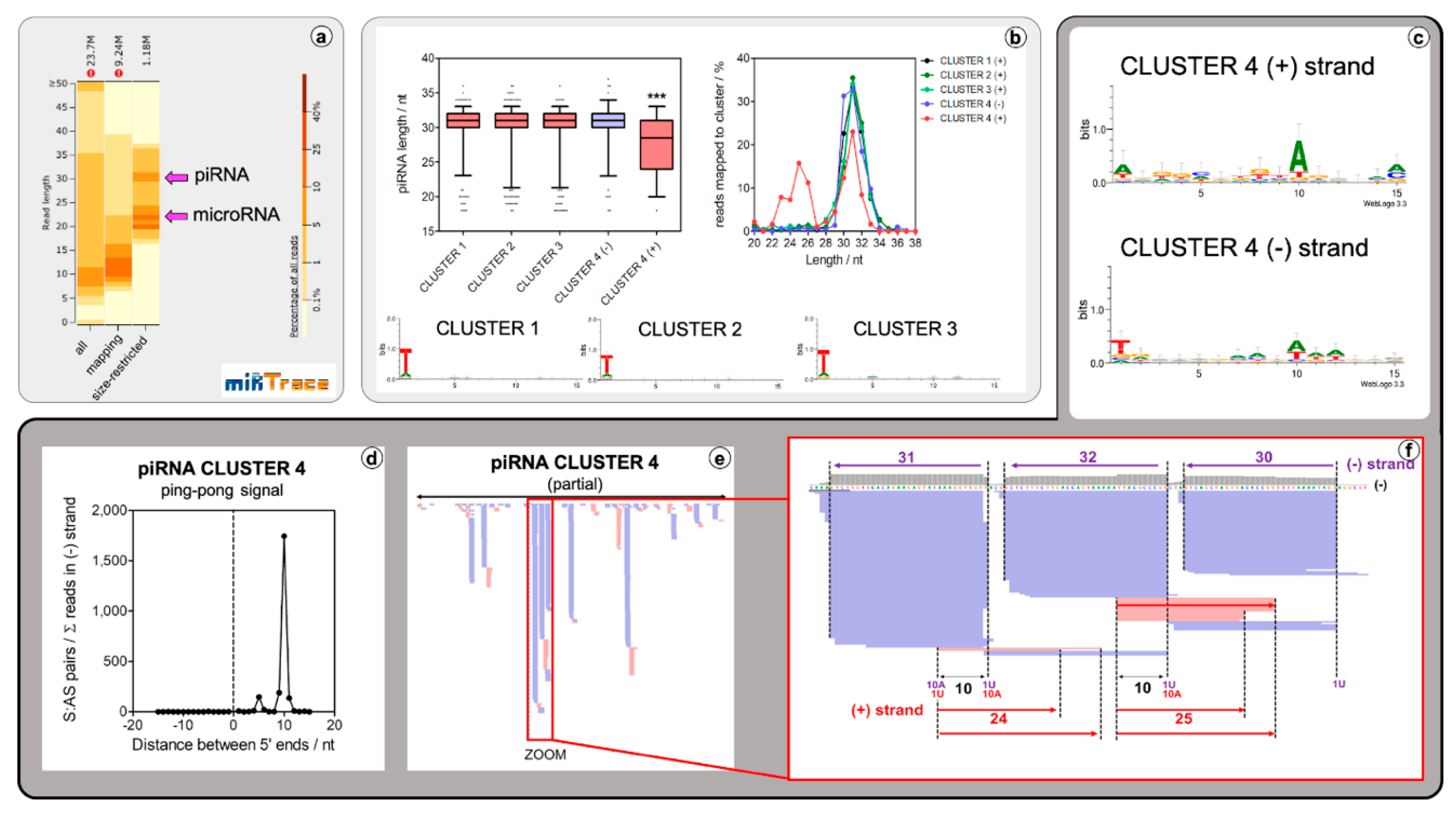
2.2. Lepidodermella squamata Has a Conventional Animal piRNA Biogenesis Mechanism
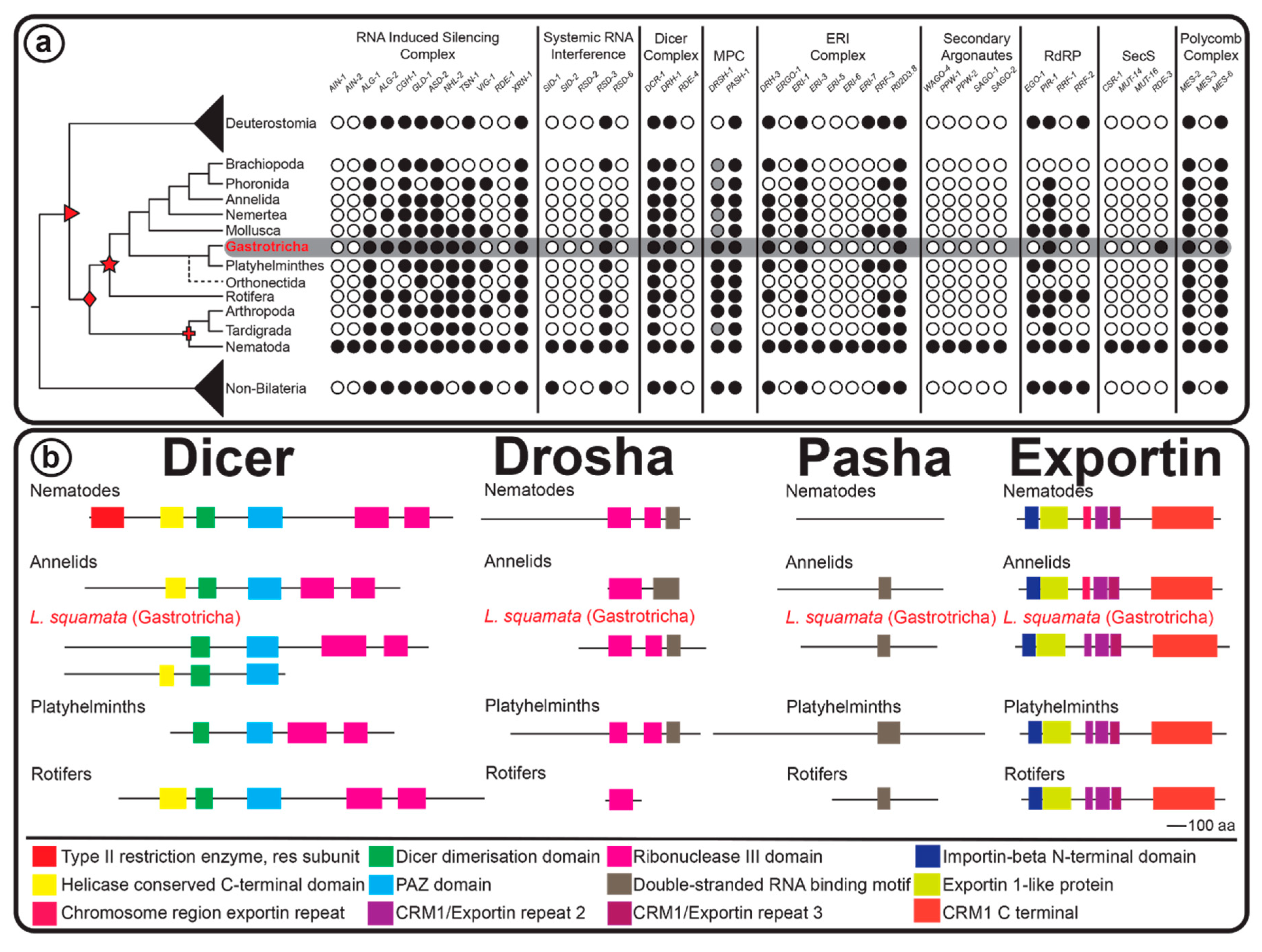
2.3. RNAi Protein Machinery of Lepidodermella squamata and Gastrotricha
3. Discussion
4. Materials and Methods
4.1. Lepidodermella squamata Culture
4.2. small-RNA Sequencing
4.3. small-RNA Bioinformatics
4.4. Identification and Domain Architecture of RNAi Proteins
4.5. Identification and Phylogenetic Analysis of PIWI Proteins
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Todaro, M.A.; Hummon, W.D. An overview and a dichotomous key to genera of the phylum Gastrotricha. Meiofauna Marina 2008, 16, 3–20. [Google Scholar]
- Balsamo, M.; d’Hondt, J.L.; Kisielewski, J.; Todaro, M.A.; Tongiorgi, P.; Guidi, L.; Grilli, P.; de Jong, Y. Fauna Europea: Gastrotricha. Biodivers. Data J. 2015, 3, e5800. [Google Scholar] [CrossRef] [PubMed]
- Paps, J.; Riutort, M. Molecular phylogeny of the phylum Gastrotricha: New data brings together molecules and morphology. Mol. Phylogenet. Evol. 2012, 63, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.A.; Telford, M.J.; Lockyer, A.E.; Littlewood, D.T.J. Interrelationships of the Gastrotricha and their place among the Metazoa inferred from 18S rRNA genes. Zool. Scr. 2006, 35, 251–259. [Google Scholar] [CrossRef]
- Hejnol, A. Gastrotricha. In Evolutionary Developmental Biology of Invertebrates 2: Lophotrochozoa (Spiralia); Wanninger, A., Ed.; Springer: Vienna, Austria, 2015; pp. 13–19. ISBN 9783709118719. [Google Scholar]
- Bekkouche, N.; Worsaae, K. Neuromuscular study of early branching Diuronotus aspetos (Paucitubulatina) yields insights into the evolution of organs systems in Gastrotricha. Zool. Lett. 2016, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.A.; Dal Zotto, M.; Leasi, F. An Integrated Morphological and Molecular Approach to the Description and Systematisation of a Novel Genus and Species of Macrodasyida (Gastrotricha). PLoS ONE 2015, 10, e0130278. [Google Scholar] [CrossRef] [PubMed]
- Rothe, B.H.; Kieneke, A.; Schmidt-Rhaesa, A. The nervous system of Xenotrichula intermedia and X. velox (Gastrotricha: Paucitubulatina) by means of immunohistochemistry (IHC) and TEM. Meiofauna Marina 2011, 19, 71–88. [Google Scholar]
- Rothe, B.H.; Schmidt-Rhaesa, A.; Kieneke, A. The nervous system of Neodasys chaetonotoideus (Gastrotricha: Neodasys) revealed by combining confocal laserscanning and transmission electron microscopy: Evolutionary comparison of neuroanatomy within the Gastrotricha and basal Protostomia. Zoomorphology 2011, 130, 51–84. [Google Scholar] [CrossRef]
- Strayer, D.L.; Hummon, W.D.; Hochberg, R. Chapter 7 - Gastrotricha. In Ecology and Classification of North American Freshwater Invertebrates (Third Edition); Thorp, J.H., Covich, A.P., Eds.; Academic Press: San Diego, CA, USA, 2010; pp. 163–172. ISBN 9780123748553. [Google Scholar]
- Zrzavy, J. Gastrotricha and metazoan phylogeny. Zool. Scr. 2003, 32, 61–81. [Google Scholar] [CrossRef]
- Hyman, L.H. The Invertebrates: Acanthocephala, Aschelminthes and Entoprocta: The Pseudocoelomate Bilateria (Volume-3); Mcgraw-Hill Book Company: New York, NY, USA; Toronto, ON, Canada; London, UK, 1951. [Google Scholar]
- Sørensen, M.V. An SEM study of the jaws of Haplognathia rosea and Rastrognathia macrostoma (Gnathostomulida), with a preliminary comparison with the rotiferan trophi. Acta Zool. 2000, 81, 9–16. [Google Scholar] [CrossRef]
- Nielsen, C. Animal Evolution: Interrelationships of the Living Phyla: Interrelationships of the Living Phyla; OUP: Oxford, UK, 2001; ISBN 9780191588525. [Google Scholar]
- Rieger, R.M. Monociliated epidermal cells in Gastrotricha: Significance for concepts of early metazoan evolution. J. Zoolog. Syst. Evol. Res. 1976, 14, 198–226. [Google Scholar] [CrossRef]
- Zrzavy, J.; Mihulka, S.; Kepka, P.; Bezdek, A.; Tietz, D. Phylogeny of the Metazoa Based on Morphological and 18S Ribosomal DNA Evidence. Cladistics 1998, 14, 249–285. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. A revised six-kingdom system of life. Biol. Rev. Camb. Philos. Soc. 1998, 73, 203–266. [Google Scholar] [CrossRef] [PubMed]
- Giribet, G. Assembling the lophotrochozoan (=spiralian) tree of life. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Edgecombe, G.; Giribet, G.; Dunn, C.; Hejnol, A.; Kristensen, R.; Neves, R.; Rouse, G.; Worsaae, K.; Sørensen, M. Higher-level metazoan relationships: recent progress and remaining questions. Org. Divers. Evol. 2011, 11, 151–172. [Google Scholar] [CrossRef]
- Struck, T.H.; Wey-Fabrizius, A.R.; Golombek, A.; Hering, L.; Weigert, A.; Bleidorn, C.; Klebow, S.; Iakovenko, N.; Hausdorf, B.; Petersen, M.; et al. Platyzoan paraphyly based on phylogenomic data supports a noncoelomate ancestry of spiralia. Mol. Biol. Evol. 2014, 31, 1833–1849. [Google Scholar] [CrossRef] [PubMed]
- Laumer, C.E.; Bekkouche, N.; Kerbl, A.; Goetz, F.; Neves, R.C.; Sørensen, M.V.; Kristensen, R.M.; Hejnol, A.; Dunn, C.W.; Giribet, G.; et al. Spiralian Phylogeny Informs the Evolution of Microscopic Lineages. Curr. Biol. 2015, 25, 2000–2006. [Google Scholar] [CrossRef] [PubMed]
- Marlétaz, F.; Peijnenburg, K.T.C.A.; Goto, T.; Satoh, N.; Rokhsar, D.S. A New Spiralian Phylogeny Places the Enigmatic Arrow Worms among Gnathiferans. Curr. Biol. 2019, 29, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Doe, D.A. Comparative ultrastructure of the pharynx simplex in turbellaria. Zoomorphology 1981, 97, 133–193. [Google Scholar] [CrossRef]
- Kieneke, A.; Riemann, O.; Ahlrichs, W.H. Novel implications for the basal internal relationships of Gastrotricha revealed by an analysis of morphological characters. Zool. Scr. 2008, 37, 429–460. [Google Scholar] [CrossRef]
- Moran, Y.; Agron, M.; Praher, D.; Technau, U. The evolutionary origin of plant and animal microRNAs. Nat. Ecol. Evol. 2017, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.W.; Siomi, M.C.; Siomi, H. PIWI-Interacting RNA: Its Biogenesis and Functions. Annu. Rev. Biochem. 2015, 84, 405–433. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Deline, B.; Greenwood, J.M.; Clark, J.W.; Puttick, M.N.; Peterson, K.J.; Donoghue, P.C.J. Evolution of metazoan morphological disparity. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [PubMed]
- Halushka, M.K.; Fromm, B.; Peterson, K.J.; McCall, M.N. Big Strides in Cellular MicroRNA Expression. Trends Genet. 2018, 34, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, F.; Raible, F.; Tomer, R.; Simakov, O.; Trachana, K.; Klaus, S.; Snyman, H.; Hannon, G.J.; Bork, P.; Arendt, D. Ancient animal microRNAs and the evolution of tissue identity. Nature 2010, 463, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, B.M.; Heimberg, A.M.; Moy, V.N.; Sperling, E.A.; Holstein, T.W.; Heber, S.; Peterson, K.J. The deep evolution of metazoan microRNAs. Evol. Dev. 2009, 11, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Tarver, J.E.; Sperling, E.A.; Nailor, A.; Heimberg, A.M.; Robinson, J.M.; King, B.L.; Pisani, D.; Donoghue, P.C.J.; Peterson, K.J. miRNAs: Small genes with big potential in metazoan phylogenetics. Mol. Biol. Evol. 2013, 30, 2369–2382. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Burow, S.; Hahn, C.; Bachmann, L. MicroRNA loci support conspecificity of Gyrodactylus salaris and Gyrodactylus thymalli (Platyhelminthes: Monogenea). Int. J. Parasitol. 2014, 44, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Kenny, N.J.; Sin, Y.W.; Hayward, A.; Paps, J.; Chu, K.H.; Hui, J.H.L. The phylogenetic utility and functional constraint of microRNA flanking sequences. Proc. Biol. Sci. 2015, 282, 20142983. [Google Scholar] [CrossRef] [PubMed]
- Helm, C.; Bernhart, S.H.; Höner zu Siederdissen, C.; Nickel, B.; Bleidorn, C. Deep sequencing of small RNAs confirms an annelid affinity of Myzostomida. Mol. Phylogenet. Evol. 2012, 64, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Billipp, T.; Peck, L.E.; Johansen, M.; Tarver, J.E.; King, B.L.; Newcomb, J.M.; Sempere, L.F.; Flatmark, K.; Hovig, E.; et al. A Uniform System for the Annotation of Vertebrate microRNA Genes and the Evolution of the Human microRNAome. Annu. Rev. Genet. 2015, 49, 213–242. [Google Scholar] [CrossRef] [PubMed]
- Tarver, J.E.; Taylor, R.S.; Puttick, M.N.; Lloyd, G.T.; Pett, W.; Fromm, B.; Schirrmeister, B.E.; Pisani, D.; Peterson, K.J.; Donoghue, P.C.J. Well-Annotated microRNAomes Do Not Evidence Pervasive miRNA Loss. Genome Biol. Evol. 2018, 10, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Thomson, R.C.; Plachetzki, D.C.; Mahler, D.L.; Moore, B.R. A critical appraisal of the use of microRNA data in phylogenetics. Proc. Natl. Acad. Sci. USA 2014, 111, E3659–E3668. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhang, Z.; Jin, L.; Kang, H.; Zhu, Y.; Zhang, L.; Li, X.; Ma, F.; Zhao, L.; Shi, B.; et al. Genome-wide sequencing of small RNAs reveals a tissue-specific loss of conserved microRNA families in Echinococcus granulosus. BMC Genom. 2014, 15, 736. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Worren, M.M.; Hahn, C.; Hovig, E.; Bachmann, L. Substantial loss of conserved and gain of novel MicroRNA families in flatworms. Mol. Biol. Evol. 2013, 30, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Philippe, H.; Brinkmann, H.; Copley, R.R.; Moroz, L.L.; Nakano, H.; Poustka, A.J.; Wallberg, A.; Peterson, K.J.; Telford, M.J. Acoelomorph flatworms are deuterostomes related to Xenoturbella. Nature 2011, 470, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, M.R.; Adamidi, C.; Ting, H.; Lebedeva, S.; Isenbarger, T.A.; Hirst, M.; Marra, M.; Nusbaum, C.; Lee, W.L.; Jenkin, J.C.; et al. High-resolution profiling and discovery of planarian small RNAs. Proc. Natl. Acad. Sci. USA 2009, 106, 11546–11551. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.J.; Zarowiecki, M.; Holroyd, N.; Garciarrubio, A.; Sanchez-Flores, A.; Brooks, K.L.; Tracey, A.; Bobes, R.J.; Fragoso, G.; Sciutto, E.; et al. The genomes of four tapeworm species reveal adaptations to parasitism. Nature 2013, 496, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Fromm, B.; Bachmann, L. Comparative genomics of flatworms (platyhelminthes) reveals shared genomic features of ecto- and endoparastic neodermata. Genome Biol. Evol. 2014, 6, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Fontenla, S.; Rinaldi, G.; Smircich, P.; Tort, J.F. Conservation and diversification of small RNA pathways within flatworms. BMC Evol. Biol. 2017, 17, 215. [Google Scholar] [CrossRef] [PubMed]
- Stroehlein, A.J.; Young, N.D.; Korhonen, P.K.; Hall, R.S.; Jex, A.R.; Webster, B.L.; Rollinson, D.; Brindley, P.J.; Gasser, R.B. The small RNA complement of adult Schistosoma haematobium. PLoS Negl. Trop. Dis. 2018, 12, e0006535. [Google Scholar] [CrossRef] [PubMed]
- Skinner, D.E.; Rinaldi, G.; Koziol, U.; Brehm, K.; Brindley, P.J. How might flukes and tapeworms maintain genome integrity without a canonical piRNA pathway? Trends Parasitol. 2014, 30, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Sarkies, P.; Selkirk, M.E.; Jones, J.T.; Blok, V.; Boothby, T.; Goldstein, B.; Hanelt, B.; Ardila-Garcia, A.; Fast, N.M.; Schiffer, P.M.; et al. Ancient and novel small RNA pathways compensate for the loss of piRNAs in multiple independent nematode lineages. PLoS Biol. 2015, 13, e1002061. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, V.Y.; Mordvinov, V.A.; Fromm, B. Extreme conservation of miRNA complements in opisthorchiids. Parasitol. Int. 2017, 66, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Ovchinnikov, V.; Høye, E.; Bernal, D.; Hackenberg, M.; Marcilla, A. On the presence and immunoregulatory functions of extracellular microRNAs in the trematode Fasciola hepatica. Parasite Immunol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Macchiaroli, N.; Cucher, M.; Zarowiecki, M.; Maldonado, L.; Kamenetzky, L.; Rosenzvit, M.C. microRNA profiling in the zoonotic parasite Echinococcus canadensis using a high-throughput approach. Parasit. Vectors 2015, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: small RNAs with big functions. Nat. Rev. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Faehnle, C.R.; Joshua-Tor, L. Argonautes confront new small RNAs. Curr. Opin. Chem. Biol. 2007, 11, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Hejnol, A.; Lowe, C.J. Embracing the comparative approach: How robust phylogenies and broader developmental sampling impacts the understanding of nervous system evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20150045. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.W.; Giribet, G.; Edgecombe, G.D.; Hejnol, A. Animal Phylogeny and Its Evolutionary Implications. Annu. Rev. Ecol. Evol. Syst. 2014, 45, 371–395. [Google Scholar] [CrossRef]
- Lu, T.-M.; Kanda, M.; Satoh, N.; Furuya, H. The phylogenetic position of dicyemid mesozoans offers insights into spiralian evolution. Zool. Lett. 2017, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Geldhof, P.; Visser, A.; Clark, D.; Saunders, G.; Britton, C.; Gilleard, J.; Berriman, M.; Knox, D. RNA interference in parasitic helminths: current situation, potential pitfalls and future prospects. Parasitology 2007, 134, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Dalzell, J.J.; McVeigh, P.; Warnock, N.D.; Mitreva, M.; Bird, D.M.; Abad, P.; Fleming, C.C.; Day, T.A.; Mousley, A.; Marks, N.J.; et al. RNAi effector diversity in nematodes. PLoS Negl. Trop. Dis. 2011, 5, e1176. [Google Scholar] [CrossRef] [PubMed]
- Büssing, I.; Yang, J.-S.; Lai, E.C.; Grosshans, H. The nuclear export receptor XPO-1 supports primary miRNA processing in C. elegans and Drosophila. EMBO J. 2010, 29, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Fontenla, S.; Dell’Oca, N.; Smircich, P.; Tort, J.F.; Siles-Lucas, M. The miRnome of Fasciola hepatica juveniles endorses the existence of a reduced set of highly divergent micro RNAs in parasitic flatworms. Int. J. Parasitol. 2015, 45, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Cucher, M.; Macchiaroli, N.; Kamenetzky, L.; Maldonado, L.; Brehm, K.; Rosenzvit, M.C. High-throughput characterization of Echinococcus spp. metacestode miRNomes. Int. J. Parasitol. 2015, 45, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.G.; Macchiaroli, N.; Lichtenstein, G.; Conti, G.; Asurmendi, S.; Milone, D.H.; Stegmayer, G.; Kamenetzky, L.; Cucher, M.; Rosenzvit, M.C. microRNA analysis of Taenia crassiceps cysticerci under praziquantel treatment and genome-wide identification of Taenia solium miRNAs. Int. J. Parasitol. 2017, 47, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Girard, A.; Sachidanandam, R.; Hannon, G.J.; Carmell, M.A. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 2006, 442, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Grivna, S.T.; Beyret, E.; Wang, Z.; Lin, H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 2006, 20, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.; Gaidatzis, D.; Pfeffer, S.; Lagos-Quintana, M.; Landgraf, P.; Iovino, N.; Morris, P.; Brownstein, M.J.; Kuramochi-Miyagawa, S.; Nakano, T.; et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 2006, 442, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-C.; Gu, W.; Shirayama, M.; Youngman, E.; Conte, D., Jr.; Mello, C.C. C. elegans piRNAs mediate the genome-wide surveillance of germline transcripts. Cell 2012, 150, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yoshikawa, M.; Han, B.W.; Izumi, N.; Tomari, Y.; Weng, Z.; Zamore, P.D. The initial uridine of primary piRNAs does not create the tenth adenine that is the hallmark of secondary piRNAs. Mol. Cell 2014, 56, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Czech, B.; Hannon, G.J. One Loop to Rule Them All: The Ping-Pong Cycle and piRNA-Guided Silencing. Trends Biochem. Sci. 2016, 41, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Jehn, J.; Gebert, D.; Pipilescu, F.; Stern, S.; Kiefer, J.S.T.; Hewel, C.; Rosenkranz, D. PIWI genes and piRNAs are ubiquitously expressed in mollusks and show patterns of lineage-specific adaptation. Commun. Biol. 2018, 1, 137. [Google Scholar] [CrossRef] [PubMed]
- Manakov, S.A.; Pezic, D.; Marinov, G.K.; Pastor, W.A.; Sachidanandam, R.; Aravin, A.A. MIWI2 and MILI Have Differential Effects on piRNA Biogenesis and DNA Methylation. Cell Rep. 2015, 12, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.H.; Quarles, K.A.; Yang, Y.; Tanguy, M.; Frézal, L.; Smith, S.A.; Sharma, P.P.; Cordaux, R.; Gilbert, C.; Giraud, I.; et al. Pan-arthropod analysis reveals somatic piRNAs as an ancestral defence against transposable elements. Nat. Ecol. Evol. 2018, 2, 174–181. [Google Scholar] [CrossRef] [PubMed]
- International Helminth Genomes Consortium Comparative genomics of the major parasitic worms. Nat. Genet. 2018.
- Weiss, M.J. Stabilization in the Name of the Freshwater Gastrotrich Lepidodermella squamata, with Nomenclatural Corrections for Congeneric Species. Trans. Am. Microsc. Soc. 1988, 107, 369–379. [Google Scholar] [CrossRef]
- Kang, W.; Eldfjell, Y.; Fromm, B.; Estivill, X.; Biryukova, I.; Friedländer, M.R. miRTrace reveals the organismal origins of microRNA sequencing data. Genome Biol. 2018, 19, 213. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, D.; Zischler, H. proTRAC—A software for probabilistic piRNA cluster detection, visualization and analysis. BMC Bioinform. 2012, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Domanska, D.; Hackenberg, M.; Mathelier, A.; Hoye, E.; Johansen, M.; Hovig, E.; Flatmark, K.; Peterson, K.J. MirGeneDB2.0: The curated microRNA Gene Database. bioRxiv 2018, 258749. [Google Scholar] [CrossRef]
- Roovers, E.F.; Rosenkranz, D.; Mahdipour, M.; Han, C.-T.; He, N.; Chuva de Sousa Lopes, S.M.; van der Westerlaken, L.A.J.; Zischler, H.; Butter, F.; Roelen, B.A.J.; et al. Piwi proteins and piRNAs in mammalian oocytes and early embryos. Cell Rep. 2015, 10, 2069–2082. [Google Scholar] [CrossRef] [PubMed]
- Tosar, J.P.; Rovira, C.; Cayota, A. Non-coding RNA fragments account for the majority of annotated piRNAs expressed in somatic non-gonadal tissues. Commun. Biol. 2018, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Schein, J.; Chiu, R.; Corbett, R.; Field, M.; Jackman, S.D.; Mungall, K.; Lee, S.; Okada, H.M.; Qian, J.Q.; et al. De novo assembly and analysis of RNA-seq data. Nat. Methods 2010, 7, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Li, G.; Liu, J.; Zhang, Y.; Ashby, C.; Liu, D.; Cramer, C.L.; Huang, X. Bridger: A new framework for de novo transcriptome assembly using RNA-seq data. Genome Biol. 2015, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Cerveau, N.; Jackson, D.J. Combining independent de novo assemblies optimizes the coding transcriptome for nonconventional model eukaryotic organisms. BMC Bioinform. 2016, 17, 525. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.-I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fromm, B.; Tosar, J.P.; Aguilera, F.; Friedländer, M.R.; Bachmann, L.; Hejnol, A. Evolutionary Implications of the microRNA- and piRNA Complement of Lepidodermella squamata (Gastrotricha). Non-Coding RNA 2019, 5, 19. https://doi.org/10.3390/ncrna5010019
Fromm B, Tosar JP, Aguilera F, Friedländer MR, Bachmann L, Hejnol A. Evolutionary Implications of the microRNA- and piRNA Complement of Lepidodermella squamata (Gastrotricha). Non-Coding RNA. 2019; 5(1):19. https://doi.org/10.3390/ncrna5010019
Chicago/Turabian StyleFromm, Bastian, Juan Pablo Tosar, Felipe Aguilera, Marc R. Friedländer, Lutz Bachmann, and Andreas Hejnol. 2019. "Evolutionary Implications of the microRNA- and piRNA Complement of Lepidodermella squamata (Gastrotricha)" Non-Coding RNA 5, no. 1: 19. https://doi.org/10.3390/ncrna5010019
APA StyleFromm, B., Tosar, J. P., Aguilera, F., Friedländer, M. R., Bachmann, L., & Hejnol, A. (2019). Evolutionary Implications of the microRNA- and piRNA Complement of Lepidodermella squamata (Gastrotricha). Non-Coding RNA, 5(1), 19. https://doi.org/10.3390/ncrna5010019