The Transcribed-Ultra Conserved Regions: Novel Non-Coding RNA Players in Neuroblastoma Progression

Abstract
:1. Neuroblastoma
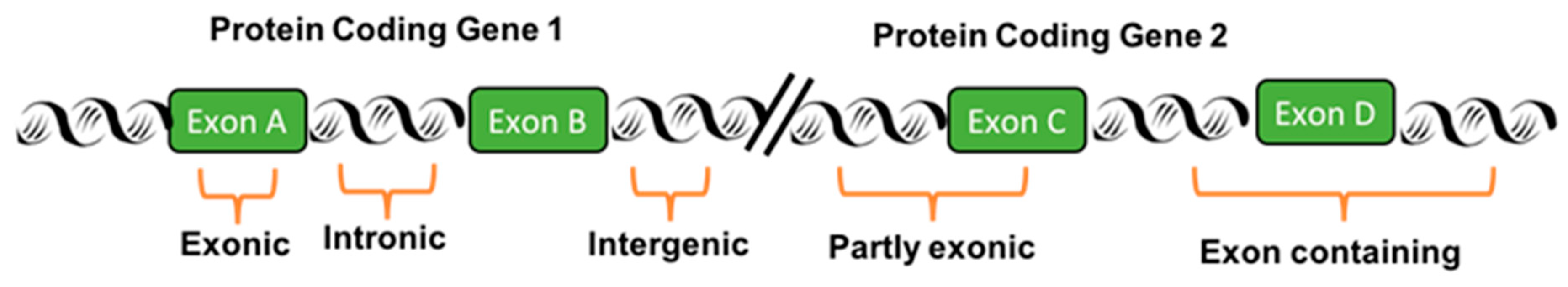
2. Transcribed-Ultra Conserved Regions
3. Regulation of Transcribed-Ultra Conserved Regions
3.1. Regulation of Gene Expression by T-UCRs
3.2. Regulation of T-UCR Expression
4. Transcribed-Ultra Conserved Regions in Neuroblastoma
4.1. T-UCRs Expression and Patient Survival in Neuroblastoma
4.2. T-UCR Expression, Genomic Locations, and Coding Genes in Neuroblastoma Tumors
4.3. T-UCR Expression and Histone Marks in Neuroblastoma
4.4. T-UCR Expression and MYCN Amplification in Neuroblastoma
4.5. T-UCR Expression & DNA Copy Number in Neuroblastoma
4.6. T-UCR Expression and p53 Response in Neuroblastoma
4.7. T-UCR Expression Network in Neuroblastoma
4.8. T-UCR Expression and Retinoic Acid Treatment
5. Transcribed-Ultra Conserved Regions in Other Cancers
5.1. T-UCRs in Hepatocellular Carcinoma (HCC)
5.2. T-UCRs in Bladder Cancer
5.3. T-UCRs in Pancreatic, Lung, Prostate, and Breast Cancers
5.4. T-UCRs in Gastric and Colon Cancers
5.5. T-UCRs in Other Cancers
6. Therapeutic Approaches in Targeting Transcribed-Ultra Conserved Regions
7. Conclusions and Future Prospective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luksch, R.; Podda, M.; Gandola, L.; Polastri, D.; Piva, L.; Castellani, R.; Collini, P.; Massimino, M.; Cefalo, G.; Terenziani, M.; et al. Stage 4 neuroblastoma: Sequential hemi-body irradiation or high-dose chemotherapy plus autologous haemopoietic stem cell transplantation to consolidate primary treatment. Br. J. Cancer 2005, 92, 1984–1988. [Google Scholar] [CrossRef] [PubMed]
- Luksch, R.; Castellani, M.R.; Collini, P.; De Bernardi, B.; Conte, M.; Gambini, C.; Gandola, L.; Garaventa, A.; Biasoni, D.; Podda, M.; et al. Neuroblastoma (Peripheral neuroblastic tumours). Crit. Rev. Oncol. Hematol. 2016, 107, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Caron, H. Allelic loss of chromosome 1 and additional chromosome 17 material are both unfavourable prognostic markers in neuroblastoma. Med. Pediatr. Oncol. 1995, 24, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Caron, H.; van Sluis, P.; de Kraker, J.; Bokkerink, J.; Egeler, M.; Laureys, G.; Slater, R.; Westerveld, A.; Voute, P.A.; Versteeg, R. Allelic loss of chromosome 1p as a predictor of unfavorable outcome in patients with neuroblastoma. N. Engl. J. Med. 1996, 334, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Pritchard, J.; Berthold, F.; Carlsen, N.L.; Castel, V.; Castelberry, R.P.; De Bernardi, B.; Evans, A.E.; Favrot, M.; Hedborg, F.; et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 1993, 11, 1466–1477. [Google Scholar] [CrossRef]
- Gatta, G.; Botta, L.; Rossi, S.; Aareleid, T.; Bielska-Lasota, M.; Clavel, J.; Dimitrova, N.; Jakab, Z.; Kaatsch, P.; Lacour, B.; et al. Childhood cancer survival in Europe 1999-2007: Results of EUROCARE-5--a population-based study. Lancet Oncol. 2014, 15, 35–47. [Google Scholar] [CrossRef]
- Trama, A.; Botta, L.; Foschi, R.; Ferrari, A.; Stiller, C.; Desandes, E.; Maule, M.M.; Merletti, F.; Gatta, G.; Group, E.-W. Survival of European adolescents and young adults diagnosed with cancer in 2000-07: Population-based data from EUROCARE-5. Lancet Oncol. 2016, 17, 896–906. [Google Scholar] [CrossRef]
- Tonini, G.P. Treatment of neuroblastoma: From cellular to molecular therapy. Curr. Pharm. Des. 2009, 15, 422–423. [Google Scholar] [CrossRef]
- De Bernardi, B.; Gambini, C.; Haupt, R.; Granata, C.; Rizzo, A.; Conte, M.; Tonini, G.P.; Bianchi, M.; Giuliano, M.; Luksch, R.; et al. Retrospective study of childhood ganglioneuroma. J. Clin. Oncol. 2008, 26, 1710–1716. [Google Scholar] [CrossRef]
- Bejerano, G.; Pheasant, M.; Makunin, I.; Stephen, S.; Kent, W.J.; Mattick, J.S.; Haussler, D. Ultraconserved elements in the human genome. Science 2004, 304, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Dickel, D.E.; Ypsilanti, A.R.; Pla, R.; Zhu, Y.; Barozzi, I.; Mannion, B.J.; Khin, Y.S.; Fukuda-Yuzawa, Y.; Plajzer-Frick, I.; Pickle, C.S.; et al. Ultraconserved Enhancers Are Required for Normal Development. Cell 2018, 172, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Mestdagh, P.; Fredlund, E.; Pattyn, F.; Rihani, A.; Van Maerken, T.; Vermeulen, J.; Kumps, C.; Menten, B.; De Preter, K.; Schramm, A.; et al. An integrative genomics screen uncovers ncRNA T-UCR functions in neuroblastoma tumours. Oncogene 2010, 29, 3583–3592. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, S.E.; Rossi, S.; Shimizu, M.; Nicoloso, M.S.; Cimmino, A.; Alder, H.; Herlea, V.; Rassenti, L.Z.; Rai, K.R.; Kipps, T.J.; et al. Non-codingRNA sequence variations in human chronic lymphocytic leukemia and colorectal cancer. Carcinogenesis 2010, 31, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Sevignani, C.; Nnadi, S.C.; Siracusa, L.D.; Calin, G.A. Cancer-associated genomic regions (CAGRs) and noncoding RNAs: Bioinformatics and therapeutic implications. Mamm. Genome 2008, 19, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Liu, C.G.; Ferracin, M.; Hyslop, T.; Spizzo, R.; Sevignani, C.; Fabbri, M.; Cimmino, A.; Lee, E.J.; Wojcik, S.E.; et al. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 2007, 12, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Portela, A.; Liz, J.; Melo, S.A.; Rossi, S.; Spizzo, R.; Croce, C.M.; Calin, G.A.; Esteller, M. CpG island hypermethylation-associated silencing of non-coding RNAs transcribed from ultraconserved regions in human cancer. Oncogene 2010, 29, 6390–6401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Wang, Z.; Zhou, J.; Liu, S.; Wu, C.; Huang, C.; Ding, Y. TUC.338 promotes invasion and metastasis in colorectal cancer. Int. J. Cancer 2017, 140, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, C.; Gong, W.; Wu, Y.; Xue, H.; Jiang, Z.; Shi, M. uc.454 Inhibited Growth by Targeting Heat Shock Protein Family A Member 12B in Non-Small-Cell Lung Cancer. Mol. Ther. Nucleic Acids 2018, 12, 174–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivieri, M.; Ferro, M.; Terreri, S.; Durso, M.; Romanelli, A.; Avitabile, C.; De Cobelli, O.; Messere, A.; Bruzzese, D.; Vannini, I.; et al. Long non-coding RNA containing ultraconserved genomic region 8 promotes bladder cancer tumorigenesis. Oncotarget 2016, 7, 20636–20654. [Google Scholar] [CrossRef]
- Nan, A.; Zhou, X.; Chen, L.; Liu, M.; Zhang, N.; Zhang, L.; Luo, Y.; Liu, Z.; Dai, L.; Jiang, Y. A transcribed ultraconserved noncoding RNA, Uc.173, is a key molecule for the inhibition of lead-induced neuronal apoptosis. Oncotarget 2016, 7, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Liz, J.; Portela, A.; Soler, M.; Gomez, A.; Ling, H.; Michlewski, G.; Calin, G.A.; Guil, S.; Esteller, M. Regulation of pri-miRNA processing by a long noncoding RNA transcribed from an ultraconserved region. Mol. Cell 2014, 55, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Ishikawa, S.; Honma, R.; Tanimoto, K.; Sakamoto, N.; Sentani, K.; Oue, N.; Teishima, J.; Matsubara, A.; Yasui, W. The transcribed-ultraconserved regions in prostate and gastric cancer: DNA hypermethylation and microRNA-associated regulation. Oncogene 2016, 35, 3598–3606. [Google Scholar] [CrossRef] [PubMed]
- Scaruffi, P.; Stigliani, S.; Moretti, S.; Coco, S.; De Vecchi, C.; Valdora, F.; Garaventa, A.; Bonassi, S.; Tonini, G.P. Transcribed-Ultra Conserved Region expression is associated with outcome in high-risk neuroblastoma. BMC Cancer 2009, 9, 441. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Esteller, M. CpG island hypermethylation of tumor suppressor microRNAs in human cancer. Cell Cycle 2007, 6, 1455–1459. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Esteller, M. How epigenetics can explain human metastasis: A new role for microRNAs. Cell Cycle 2009, 8, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.S.; Yi, M.; Volfovsky, N.; Prueitt, R.L.; Esposito, D.; Volinia, S.; Liu, C.G.; Schetter, A.J.; Van Roosbroeck, K.; Stephens, R.M.; et al. Transcription signatures encoded by ultraconserved genomic regions in human prostate cancer. Mol. Cancer 2013, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, D.L.; Pandit, K.V.; Gordon, B.; Bhattacharjee, A.; Kaminski, N.; Benos, P.V. Features of mammalian microRNA promoters emerge from polymerase II chromatin immunoprecipitation data. PLoS ONE 2009, 4, 5279. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef]
- Ferdin, J.; Nishida, N.; Wu, X.; Nicoloso, M.S.; Shah, M.Y.; Devlin, C.; Ling, H.; Shimizu, M.; Kumar, K.; Cortez, M.A.; et al. HINCUTs in cancer: Hypoxia-induced noncoding ultraconserved transcripts. Cell Death Differ. 2013, 20, 1675–1687. [Google Scholar] [CrossRef]
- Vermeulen, J.; De Preter, K.; Naranjo, A.; Vercruysse, L.; Van Roy, N.; Hellemans, J.; Swerts, K.; Bravo, S.; Scaruffi, P.; Tonini, G.P.; et al. Predicting outcomes for children with neuroblastoma using a multigene-expression signature: A retrospective SIOPEN/COG/GPOH study. Lancet Oncol. 2009, 10, 663–671. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Kamal, M.; Lindblad-Toh, K.; Bekiranov, S.; Bailey, D.K.; Huebert, D.J.; McMahon, S.; Karlsson, E.K.; Kulbokas, E.J., 3rd; Gingeras, T.R.; et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 2005, 120, 169–181. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Schulte, J.H.; Horn, S.; Otto, T.; Samans, B.; Heukamp, L.C.; Eilers, U.C.; Krause, M.; Astrahantseff, K.; Klein-Hitpass, L.; Buettner, R.; et al. MYCN regulates oncogenic MicroRNAs in neuroblastoma. Int. J. Cancer 2008, 122, 699–704. [Google Scholar] [CrossRef]
- Depuydt, P.; Boeva, V.; Hocking, T.D.; Cannoodt, R.; Ambros, I.M.; Ambros, P.F.; Asgharzadeh, S.; Attiyeh, E.F.; Combaret, V.; Defferrari, R.; et al. Genomic Amplifications and Distal 6q Loss: Novel Markers for Poor Survival in High-risk Neuroblastoma Patients. J. Natl. Cancer Inst. 2018, 110, 1084–1093. [Google Scholar] [CrossRef]
- Powers, J.T.; Tsanov, K.M.; Pearson, D.S.; Roels, F.; Spina, C.S.; Ebright, R.; Seligson, M.; de Soysa, Y.; Cahan, P.; Theissen, J.; et al. Multiple mechanisms disrupt the let-7 microRNA family in neuroblastoma. Nature 2016, 535, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Sanmartin, E.; Munoz, L.; Piqueras, M.; Sirerol, J.A.; Berlanga, P.; Canete, A.; Castel, V.; Font de Mora, J. Deletion of 11q in Neuroblastomas Drives Sensitivity to PARP Inhibition. Clin. Cancer Res. 2017, 23, 6875–6887. [Google Scholar] [CrossRef]
- Vandesompele, J.; Baudis, M.; De Preter, K.; Van Roy, N.; Ambros, P.; Bown, N.; Brinkschmidt, C.; Christiansen, H.; Combaret, V.; Lastowska, M.; et al. Unequivocal delineation of clinicogenetic subgroups and development of a new model for improved outcome prediction in neuroblastoma. J. Clin. Oncol. 2005, 23, 2280–2299. [Google Scholar] [CrossRef]
- Lazcoz, P.; Munoz, J.; Nistal, M.; Pestana, A.; Encio, I.J.; Castresana, J.S. Loss of heterozygosity and microsatellite instability on chromosome arm 10q in neuroblastoma. Cancer Genet. Cytogenet. 2007, 174, 1–8. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Van Maerken, T.; Speleman, F.; Vermeulen, J.; Lambertz, I.; De Clercq, S.; De Smet, E.; Yigit, N.; Coppens, V.; Philippe, J.; De Paepe, A.; et al. Small-molecule MDM2 antagonists as a new therapy concept for neuroblastoma. Cancer Res. 2006, 66, 9646–9655. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Watters, K.M.; Bryan, K.; Foley, N.H.; Meehan, M.; Stallings, R.L. Expressional alterations in functional ultra-conserved non-coding RNAs in response to all-trans retinoic acid--induced differentiation in neuroblastoma cells. BMC Cancer 2013, 13, 184. [Google Scholar] [CrossRef]
- Braconi, C.; Valeri, N.; Kogure, T.; Gasparini, P.; Huang, N.; Nuovo, G.J.; Terracciano, L.; Croce, C.M.; Patel, T. Expression and functional role of a transcribed noncoding RNA with an ultraconserved element in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 786–791. [Google Scholar] [CrossRef]
- Kogure, T.; Yan, I.K.; Lin, W.L.; Patel, T. Extracellular Vesicle-Mediated Transfer of a Novel Long Noncoding RNA TUC339: A Mechanism of Intercellular Signaling in Human Hepatocellular Cancer. Genes Cancer 2013, 4, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Carotenuto, P.; Fassan, M.; Pandolfo, R.; Lampis, A.; Vicentini, C.; Cascione, L.; Paulus-Hock, V.; Boulter, L.; Guest, R.; Quagliata, L.; et al. Wnt signalling modulates transcribed-ultraconserved regions in hepatobiliary cancers. Gut 2017, 66, 1268–1277. [Google Scholar] [CrossRef]
- Luo, H.L.; Chen, J.; Luo, T.; Wu, F.X.; Liu, J.J.; Wang, H.F.; Chen, M.; Li, L.Q.; Li, H. Downregulation of Macrophage-Derived T-UCR uc.306 Associates with Poor Prognosis in Hepatocellular Carcinoma. Cell Physiol. Biochem. 2017, 42, 1526–1539. [Google Scholar] [CrossRef] [Green Version]
- Terreri, S.; Durso, M.; Colonna, V.; Romanelli, A.; Terracciano, D.; Ferro, M.; Perdona, S.; Castaldo, L.; Febbraio, F.; de Nigris, F.; et al. New Cross-Talk Layer between Ultraconserved Non-Coding RNAs, MicroRNAs and Polycomb Protein YY1 in Bladder Cancer. Genes 2016, 7, 127. [Google Scholar] [CrossRef]
- Jiang, J.; Azevedo-Pouly, A.C.; Redis, R.S.; Lee, E.J.; Gusev, Y.; Allard, D.; Sutaria, D.S.; Badawi, M.; Elgamal, O.A.; Lerner, M.R.; et al. Globally increased ultraconserved noncoding RNA expression in pancreatic adenocarcinoma. Oncotarget 2016, 7, 53165–53177. [Google Scholar] [CrossRef]
- Vannini, I.; Wise, P.M.; Challagundla, K.B.; Plousiou, M.; Raffini, M.; Bandini, E.; Fanini, F.; Paliaga, G.; Crawford, M.; Ferracin, M.; et al. Transcribed ultraconserved region 339 promotes carcinogenesis by modulating tumor suppressor microRNAs. Nat. Commun. 2017, 8, 1801. [Google Scholar] [CrossRef]
- Sekino, Y.; Sakamoto, N.; Goto, K.; Honma, R.; Shigematsu, Y.; Sentani, K.; Oue, N.; Teishima, J.; Matsubara, A.; Yasui, W. Transcribed ultraconserved region Uc.63+ promotes resistance to docetaxel through regulation of androgen receptor signaling in prostate cancer. Oncotarget 2017, 8, 94259–94270. [Google Scholar] [CrossRef]
- Marini, A.; Lena, A.M.; Panatta, E.; Ivan, C.; Han, L.; Liang, H.; Annicchiarico-Petruzzelli, M.; Di Daniele, N.; Calin, G.A.; Candi, E.; et al. Ultraconserved long non-coding RNA uc.63 in breast cancer. Oncotarget 2017, 8, 35669–35680. [Google Scholar] [CrossRef]
- Honma, R.; Goto, K.; Sakamoto, N.; Sekino, Y.; Sentani, K.; Oue, N.; Yasui, W. Expression and function of Uc.160+, a transcribed ultraconserved region, in gastric cancer. Gastric Cancer 2017, 20, 960–969. [Google Scholar] [CrossRef]
- Qian, X.X.; Peng, J.C.; Xu, A.T.; Zhao, D.; Qiao, Y.Q.; Wang, T.R.; Shen, J.; Ran, Z.H. Noncoding Transcribed Ultraconserved Region (T-UCR) uc.261 Participates in Intestinal Mucosa Barrier Damage in Crohn’s Disease. Inflamm. Bowel Dis. 2016, 22, 2840–2852. [Google Scholar] [CrossRef]
- Pang, L.; Li, Q.; Zhang, Y.; Deng, B.; Wu, F.; Wang, J.; Wu, K.; Ding, Y.; Yu, D. Transcribed ultraconserved noncoding RNA uc.160 acts as a negative regulator in gastric cancer. Am. J. Transl. Res. 2018, 10, 2822–2833. [Google Scholar]
- Xiao, L.; Wu, J.; Wang, J.Y.; Chung, H.K.; Kalakonda, S.; Rao, J.N.; Gorospe, M.; Wang, J.Y. Long Noncoding RNA uc.173 Promotes Renewal of the Intestinal Mucosa by Inducing Degradation of MicroRNA 195. Gastroenterology 2018, 154, 599–611. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Y.; Cui, Y.H.; Xiao, L.; Chung, H.K.; Zhang, Y.; Rao, J.N.; Gorospe, M.; Wang, J.Y. Regulation of Intestinal Epithelial Barrier Function by Long Noncoding RNA uc.173 through Interaction with MicroRNA 29b. Mol. Cell. Biol. 2018, 38, 10–18. [Google Scholar] [CrossRef]
- Kottorou, A.E.; Antonacopoulou, A.G.; Dimitrakopoulos, F.D.; Diamantopoulou, G.; Sirinian, C.; Kalofonou, M.; Theodorakopoulos, T.; Oikonomou, C.; Katsakoulis, E.C.; Koutras, A.; et al. Deregulation of methylation of transcribed-ultra conserved regions in colorectal cancer and their value for detection of adenomas and adenocarcinomas. Oncotarget 2018, 9, 21411–21428. [Google Scholar] [CrossRef] [Green Version]
- Sana, J.; Hankeova, S.; Svoboda, M.; Kiss, I.; Vyzula, R.; Slaby, O. Expression levels of transcribed ultraconserved regions uc.73 and uc.388 are altered in colorectal cancer. Oncology 2012, 82, 114–118. [Google Scholar] [CrossRef]
- Galasso, M.; Dama, P.; Previati, M.; Sandhu, S.; Palatini, J.; Coppola, V.; Warner, S.; Sana, M.E.; Zanella, R.; Abujarour, R.; et al. A large scale expression study associates uc.283-plus lncRNA with pluripotent stem cells and human glioma. Genome Med. 2014, 6, 76. [Google Scholar] [CrossRef]
- Fassan, M.; Dall’Olmo, L.; Galasso, M.; Braconi, C.; Pizzi, M.; Realdon, S.; Volinia, S.; Valeri, N.; Gasparini, P.; Baffa, R.; et al. Transcribed ultraconserved noncoding RNAs (T-UCR) are involved in Barrett’s esophagus carcinogenesis. Oncotarget 2014, 5, 7162–7171. [Google Scholar] [CrossRef]
- Zhang, L.X.; Xu, L.; Zhang, C.H.; Lu, Y.H.; Ji, T.H.; Ling, L.J. uc.38 induces breast cancer cell apoptosis via PBX1. Am. J. Cancer Res. 2017, 7, 2438–2451. [Google Scholar]
- Zhou, J.; Wang, R.; Zhang, J.; Zhu, L.; Liu, W.; Lu, S.; Chen, P.; Li, H.; Yin, B.; Yuan, J.; et al. Conserved expression of ultra-conserved noncoding RNA in mammalian nervous system. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 1159–1168. [Google Scholar] [CrossRef]
- Sekino, Y.; Sakamoto, N.; Goto, K.; Honma, R.; Shigematsu, Y.; Quoc, T.P.; Sentani, K.; Oue, N.; Teishima, J.; Kawakami, F.; et al. Uc.416 + A promotes epithelial-to-mesenchymal transition through miR-153 in renal cell carcinoma. BMC Cancer 2018, 18, 952. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| T-UCR Name | Chromosome Location | Start (bp) | End (bp) | T-UCR Expression | miRNA | miRNA Expression |
|---|---|---|---|---|---|---|
| uc.209 | 7 | 23,561,888 | 23,562,137 | ↑ | hsa-miR-877-3p | ↓ |
| uc.271 | 9 | 128,304,352 | 128,304,562 | ↑ | hsa-miR-383 | ↓ |
| uc.312 | 10 | 120,076,537 | 120,076,858 | ↑ | hsa-miR-877-3p, hsa-miR-548d-5p | ↓ |
| uc.330 | 11 | 66,393,896 | 66,394,102 | ↑ | hsa-miR-548d-5p | ↓ |
| uc.371 | 14 | 36,020,189 | 36,020,484 | ↑ | hsa-miR-877-3p | ↓ |
| uc.411 | 17 | 35,329,619 | 35,329,847 | ↑ | hsa-miR-33b-5p | ↓ |
| uc.421 | 18 | 22,693,155 | 22,693,499 | ↑ | hsa-miR-877-3p | ↓ |
| uc.435 | 18 | 53,089,931 | 53,090,157 | ↑ | hsa-miR-939 | ↓ |
| uc.452 | 19 | 31,827,947 | 31,828,150 | ↑ | hsa-miR-383 | ↓ |
| T-UCR | Location | Start (bp) | End (bp) |
|---|---|---|---|
| uc.10 | 1 | 10,965,574 | 10,965,848 |
| uc.25 | 1 | 51,166,034 | 51,166,268 |
| uc.300 | 10 | 102,547,118 | 102,547,325 |
| uc.303 | 10 | 103,052,427 | 103,052,698 |
| uc.308 | 10 | 103,245,812 | 103,246,088 |
| uc.379 | 14 | 97,431,368 | 97,431,619 |
| uc.380 | 14 | 97,762,594 | 97,762,825 |
| Cluster | Pathway | T-UCR | Chromosome Location |
|---|---|---|---|
| Cluster 1 | uc.31 | 1 | |
| uc.58 | 2 | ||
| DNA | uc.130 | 3 | |
| Damage | uc.139 | 4 | |
| Response | uc.196 | 6 | |
| uc.293 | 10 | ||
| uc.296 | 10 | ||
| uc.365 | 14 | ||
| uc.405 | 16 | ||
| Cluster 2 | uc.74 | 2 | |
| uc.103 | 2 | ||
| uc.104 | 2 | ||
| uc.131 | 3 | ||
| uc.134 | 3 | ||
| Cell Cycle and | uc.257 | 9 | |
| Proliferation | uc.277 | 9 | |
| uc.278 | 9 | ||
| uc.279 | 9 | ||
| uc.431 | 18 | ||
| uc.444 | 19 | ||
| uc.483 | 3 | ||
| Cluster 3 | uc.16 | 1 | |
| uc.30 | 1 | ||
| uc.46 | 1 | ||
| uc.49 | 2 | ||
| Differentiation | uc.101 | 2 | |
| uc.193 | 6 | ||
| uc.366 | 14 | ||
| uc.380 | 14 | ||
| uc.456 | 20 | ||
| Cluster 4 | uc.21 | 1 | |
| uc.65 | 2 | ||
| Immune | uc.98 | 2 | |
| Response and | uc.145 | 4 | |
| Development | uc.334 | 11 | |
| uc.347 | 13 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mudgapalli, N.; Shaw, B.P.; Chava, S.; Challagundla, K.B. The Transcribed-Ultra Conserved Regions: Novel Non-Coding RNA Players in Neuroblastoma Progression. Non-Coding RNA 2019, 5, 39. https://doi.org/10.3390/ncrna5020039
Mudgapalli N, Shaw BP, Chava S, Challagundla KB. The Transcribed-Ultra Conserved Regions: Novel Non-Coding RNA Players in Neuroblastoma Progression. Non-Coding RNA. 2019; 5(2):39. https://doi.org/10.3390/ncrna5020039
Chicago/Turabian StyleMudgapalli, Nithya, Brianna P. Shaw, Srinivas Chava, and Kishore B. Challagundla. 2019. "The Transcribed-Ultra Conserved Regions: Novel Non-Coding RNA Players in Neuroblastoma Progression" Non-Coding RNA 5, no. 2: 39. https://doi.org/10.3390/ncrna5020039
APA StyleMudgapalli, N., Shaw, B. P., Chava, S., & Challagundla, K. B. (2019). The Transcribed-Ultra Conserved Regions: Novel Non-Coding RNA Players in Neuroblastoma Progression. Non-Coding RNA, 5(2), 39. https://doi.org/10.3390/ncrna5020039