RNA-Centric Approaches to Profile the RNA–Protein Interaction Landscape on Selected RNAs


Abstract
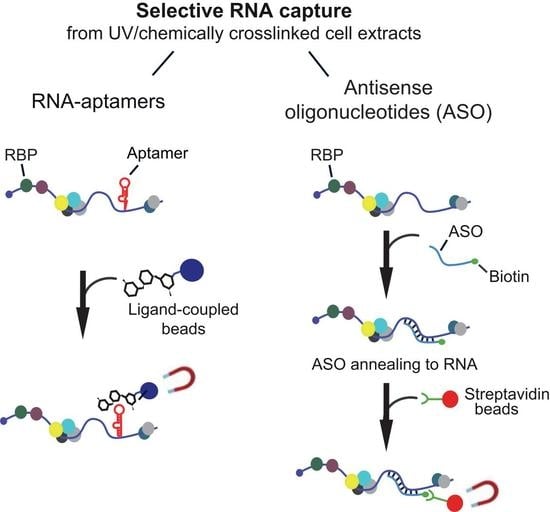
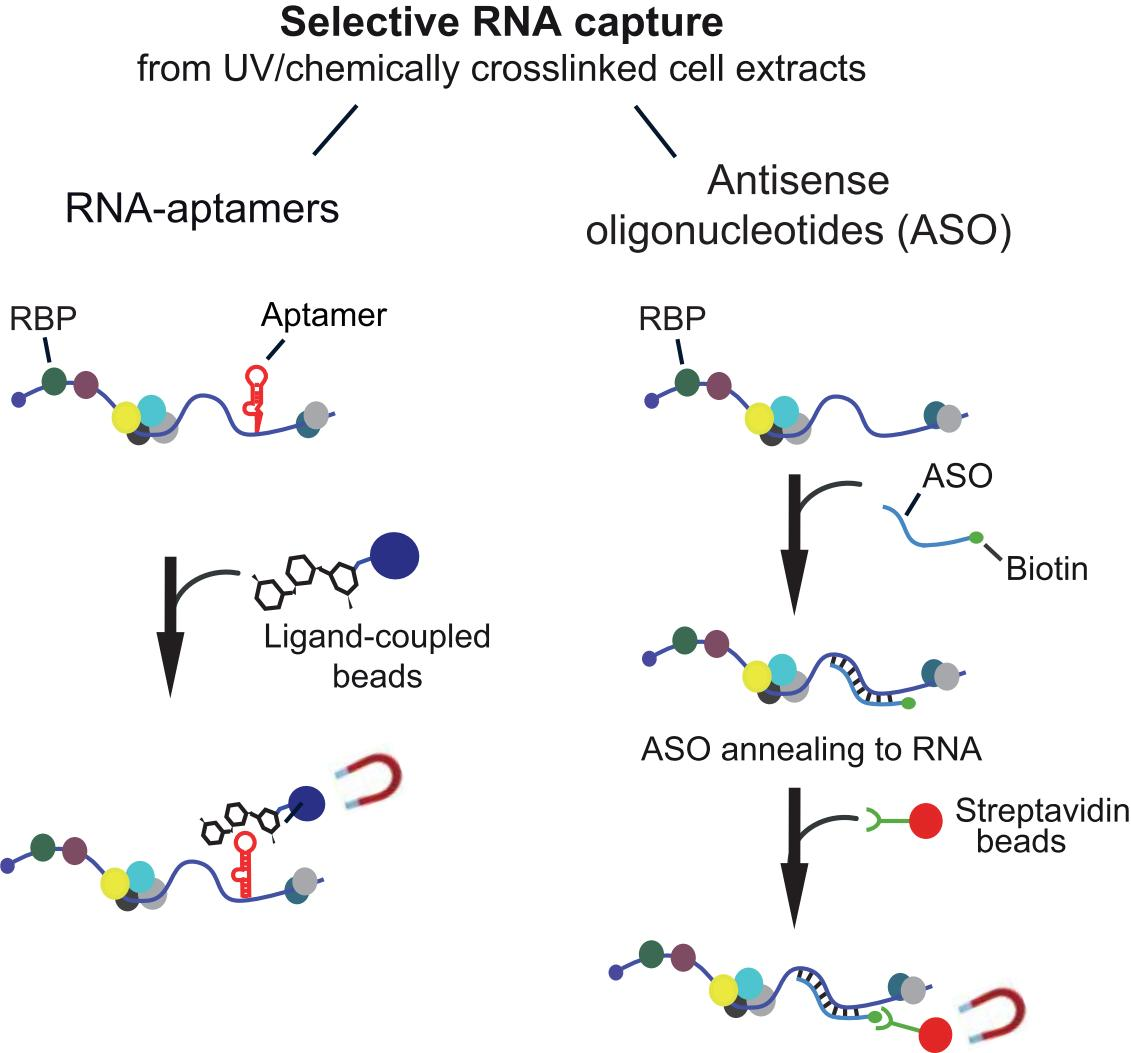
:
1. Introduction
2. RNA-Centric Approaches to Capture Selected RNAs—Overview and Concepts
3. Affinity Capture of RNAs via Aptamers
4. Capturing Endogenous RNAs with ASOs
4.1. Capture of lncRNAs with ASOs
4.2. Capture of Viral RNAs
4.3. Specific mRNA Capture
5. CRISPR-Cas Based In Vivo Targeting Approaches
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Uszczynska-Ratajczak, B.; Lagarde, J.; Frankish, A.; Guigo, R.; Johnson, R. Towards a complete map of the human long non-coding RNA transcriptome. Nat. Rev. Genet. 2018, 19, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Anantharaman, V.; Koonin, E.V.; Aravind, L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002, 30, 1427–1464. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, F.; Schwarzl, T.; Valcarcel, J.; Hentze, M.W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2020, 416–425. [Google Scholar] [CrossRef]
- Iadevaia, V.; Gerber, A.P. Combinatorial Control of mRNA Fates by RNA-Binding Proteins and Non-Coding RNAs. Biomolecules 2015, 5, 2207–2222. [Google Scholar] [CrossRef] [Green Version]
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Corley, M.; Burns, M.C.; Yeo, G.W. How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol. Cell 2020, 78, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Albihlal, W.S.; Gerber, A.P. Unconventional RNA-binding proteins: An uncharted zone in RNA biology. FEBS Lett. 2018, 592, 2917–2931. [Google Scholar] [CrossRef]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Tenenbaum, S.A.; Carson, C.C.; Lager, P.J.; Keene, J.D. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc. Natl. Acad. Sci. USA 2000, 97, 14085–14090. [Google Scholar] [CrossRef] [Green Version]
- Gerber, A.P.; Herschlag, D.; Brown, P.O. Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol. 2004, 2, E79. [Google Scholar] [CrossRef]
- Nicholson, C.O.; Friedersdorf, M.; Keene, J.D. Quantifying RNA binding sites transcriptome-wide using DO-RIP-seq. RNA 2017, 23, 32–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, F.C.Y.; Ule, J. Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Mol. Cell 2018, 69, 354–369. [Google Scholar] [CrossRef] [Green Version]
- Hogan, D.J.; Riordan, D.P.; Gerber, A.P.; Herschlag, D.; Brown, P.O. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008, 6, e255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.Y.; Cody, N.A.L.; Dominguez, D.; et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 2020, 583, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Keene, J.D. RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.; Kanitz, A.; Gerber, A.P. RNA regulons and the RNA-protein interaction network. Biomol. Concepts 2012, 3, 403–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherrer, T.; Mittal, N.; Janga, S.C.; Gerber, A.P. A screen for RNA-binding proteins in yeast indicates dual functions for many enzymes. PLoS ONE 2010, 5, e15499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsvetanova, N.G.; Klass, D.M.; Salzman, J.; Brown, P.O. Proteome-wide search reveals unexpected RNA-binding proteins in Saccharomyces cerevisiae. PLoS ONE 2010, 5, e12671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltz, A.G.; Munschauer, M.; Schwanhausser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef] [Green Version]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckmann, B.M.; Horos, R.; Fischer, B.; Castello, A.; Eichelbaum, K.; Alleaume, A.M.; Schwarzl, T.; Curk, T.; Foehr, S.; Huber, W.; et al. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun. 2015, 6, 10127. [Google Scholar] [CrossRef] [PubMed]
- Matia-Gonzalez, A.M.; Laing, E.E.; Gerber, A.P. Conserved mRNA-binding proteomes in eukaryotic organisms. Nat. Struct. Mol. Biol. 2015, 22, 1027–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Perri, J.I.; Rogell, B.; Schwarzl, T.; Stein, F.; Zhou, Y.; Rettel, M.; Brosig, A.; Hentze, M.W. Discovery of RNA-binding proteins and characterization of their dynamic responses by enhanced RNA interactome capture. Nat. Commun. 2018, 9, 4408. [Google Scholar] [CrossRef] [Green Version]
- Queiroz, R.M.L.; Smith, T.; Villanueva, E.; Marti-Solano, M.; Monti, M.; Pizzinga, M.; Mirea, D.M.; Ramakrishna, M.; Harvey, R.F.; Dezi, V.; et al. Comprehensive identification of RNA-protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat. Biotechnol. 2019, 37, 169–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urdaneta, E.C.; Vieira-Vieira, C.H.; Hick, T.; Wessels, H.H.; Figini, D.; Moschall, R.; Medenbach, J.; Ohler, U.; Granneman, S.; Selbach, M.; et al. Purification of cross-linked RNA-protein complexes by phenol-toluol extraction. Nat. Commun. 2019, 10, 990. [Google Scholar] [CrossRef]
- Trendel, J.; Schwarzl, T.; Horos, R.; Prakash, A.; Bateman, A.; Hentze, M.W.; Krijgsveld, J. The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 2019, 176, 391–403.e19. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Guo, X.; Yin, M.; Tariq, M.; Lai, Y.; Kanwal, S.; Zhou, J.; Li, N.; Lv, Y.; Pulido-Quetglas, C.; et al. Capturing the interactome of newly transcribed RNA. Nat. Methods 2018, 15, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Han, M.; Meng, L.; Chen, X. Transcriptome-wide discovery of coding and noncoding RNA-binding proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E3879–E3887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castello, A.; Fischer, B.; Frese, C.K.; Horos, R.; Alleaume, A.M.; Foehr, S.; Curk, T.; Krijgsveld, J.; Hentze, M.W. Comprehensive Identification of RNA-Binding Domains in Human Cells. Mol. Cell 2016, 63, 696–710. [Google Scholar] [CrossRef] [Green Version]
- Shchepachev, V.; Bresson, S.; Spanos, C.; Petfalski, E.; Fischer, L.; Rappsilber, J.; Tollervey, D. Defining the RNA interactome by total RNA-associated protein purification. Mol. Syst. Biol. 2019, 15, e8689. [Google Scholar] [CrossRef]
- Bae, J.W.; Kwon, S.C.; Na, Y.; Kim, V.N.; Kim, J.S. Chemical RNA digestion enables robust RNA-binding site mapping at single amino acid resolution. Nat. Struct. Mol. Biol. 2020, 27, 678–682. [Google Scholar] [CrossRef]
- Ramanathan, M.; Porter, D.F.; Khavari, P.A. Methods to study RNA-protein interactions. Nat. Methods 2019, 16, 225–234. [Google Scholar] [CrossRef]
- Van Ende, R.; Balzarini, S.; Geuten, K. Single and Combined Methods to Specifically or Bulk-Purify RNA-Protein Complexes. Biomolecules 2020, 10, 1160. [Google Scholar] [CrossRef]
- Dasti, A.; Cid-Samper, F.; Bechara, E.; Tartaglia, G.G. RNA-centric approaches to study RNA-protein interactions in vitro and in silico. Methods 2020, 178, 11–18. [Google Scholar] [CrossRef]
- Strehle, M.; Guttman, M. Xist drives spatial compartmentalization of DNA and protein to orchestrate initiation and maintenance of X inactivation. Curr. Opin. Cell Biol. 2020, 64, 139–147. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Wang, X.; Podell, E.R.; Cech, T.R. RNA seeds higher-order assembly of FUS protein. Cell Rep. 2013, 5, 918–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, K.; Prandl, R.; Schoffl, F. Detecting DNA-binding of proteins in vivo by UV-crosslinking and immunoprecipitation. Biochem. Biophys. Res. Commun. 2004, 322, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Gemmill, D.; D’Souza, S.; Meier-Stephenson, V.; Patel, T.R. Current approaches for RNA-labelling to identify RNA-binding proteins. Biochem. Cell Biol. 2020, 98, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, K.; Killian, J.; Cho, J.; Rando, R.R. Minimal RNA constructs that specifically bind aminoglycoside antibiotics with high affinities. Biochemistry 1998, 37, 656–663. [Google Scholar] [CrossRef]
- Bachler, M.; Schroeder, R.; von Ahsen, U. StreptoTag: A novel method for the isolation of RNA-binding proteins. RNA 1999, 5, 1509–1516. [Google Scholar] [CrossRef] [Green Version]
- Hartmuth, K.; Vornlocher, H.P.; Luhrmann, R. Tobramycin affinity tag purification of spliceosomes. Methods Mol. Biol. 2004, 257, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Pianzola, P.; Urlaub, H.; Rivera-Pomar, R. Proteomic analysis of reaper 5’ untranslated region-interacting factors isolated by tobramycin affinity-selection reveals a role for La antigen in reaper mRNA translation. Proteomics 2005, 5, 1645–1655. [Google Scholar] [CrossRef]
- Windbichler, N.; Schroeder, R. Isolation of specific RNA-binding proteins using the streptomycin-binding RNA aptamer. Nat. Protoc. 2006, 1, 637–640. [Google Scholar] [CrossRef]
- Iadevaia, V.; Wouters, M.D.; Kanitz, A.; Matia-Gonzalez, A.M.; Laing, E.E.; Gerber, A.P. Tandem RNA isolation reveals functional rearrangement of RNA-binding proteins on CDKN1B/p27(Kip1) 3’UTRs in cisplatin treated cells. RNA Biol. 2020, 17, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Slobodin, B.; Gerst, J.E. A novel mRNA affinity purification technique for the identification of interacting proteins and transcripts in ribonucleoprotein complexes. RNA 2010, 16, 2277–2290. [Google Scholar] [CrossRef] [Green Version]
- Leppek, K.; Stoecklin, G. An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res. 2014, 42, e13. [Google Scholar] [CrossRef] [Green Version]
- Hogg, J.R.; Collins, K. RNA-based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA 2007, 13, 868–880. [Google Scholar] [CrossRef] [Green Version]
- Di Tomasso, G.; Jenkins, L.M.M.; Legault, P. ARiBo pull-down for riboproteomic studies based on label-free quantitative mass spectrometry. RNA 2016, 22, 1760–1770. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Haurwitz, R.E.; Apffel, A.; Zhou, K.; Smart, B.; Wenger, C.D.; Laderman, S.; Bruhn, L.; Doudna, J.A. RNA-protein analysis using a conditional CRISPR nuclease. Proc. Natl. Acad. Sci. USA 2013, 110, 5416–5421. [Google Scholar] [CrossRef] [Green Version]
- McDermott, J.J.; Watkins, K.P.; Williams-Carrier, R.; Barkan, A. Ribonucleoprotein Capture by in Vivo Expression of a Designer Pentatricopeptide Repeat Protein in Arabidopsis. Plant Cell 2019, 31, 1723–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, M.; Levin, M.; Butter, F.; Scheibe, M. Quantitative Proteomics to Identify Nuclear RNA-Binding Proteins of Malat1. Int. J. Mol. Sci. 2020, 21, 1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faoro, C.; Ataide, S.F. Ribonomic approaches to study the RNA-binding proteome. FEBS Lett. 2014, 588, 3649–3664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, J.R.; Goff, S.P. Upf1 senses 3’UTR length to potentiate mRNA decay. Cell 2010, 143, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, B.P.; Wang, X.; Huang, L.; Waterman, M.L. Quantitative profiling of in vivo-assembled RNA-protein complexes using a novel integrated proteomic approach. Mol. Cell. Proteom. 2011, 10, M110.007385. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Gorospe, M. Identification of mRNA-Interacting Factors by MS2-TRAP (MS2-Tagged RNA Affinity Purification). Methods Mol. Biol. 2016, 1421, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, M.; Majzoub, K.; Rao, D.S.; Neela, P.H.; Zarnegar, B.J.; Mondal, S.; Roth, J.G.; Gai, H.; Kovalski, J.R.; Siprashvili, Z.; et al. RNA-protein interaction detection in living cells. Nat. Methods 2018, 15, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Blencowe, B.J.; Sproat, B.S.; Ryder, U.; Barabino, S.; Lamond, A.I. Antisense probing of the human U4/U6 snRNP with biotinylated 2’-OMe RNA oligonucleotides. Cell 1989, 59, 531–539. [Google Scholar] [CrossRef]
- Lingner, J.; Cech, T.R. Purification of telomerase from Euplotes aediculatus: Requirement of a primer 3’ overhang. Proc. Natl. Acad. Sci. USA 1996, 93, 10712–10717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnapp, G.; Rodi, H.P.; Rettig, W.J.; Schnapp, A.; Damm, K. One-step affinity purification protocol for human telomerase. Nucleic Acids Res. 1998, 26, 3311–3313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.; Zhang, Q.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic discovery of Xist RNA binding proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Piao, H.L.; Kim, B.J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Minajigi, A.; Froberg, J.; Wei, C.; Sunwoo, H.; Kesner, B.; Colognori, D.; Lessing, D.; Payer, B.; Boukhali, M.; Haas, W.; et al. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiniello, M.; Knoener, R.A.; Steinbrink, M.I.; Yang, B.; Cesnik, A.J.; Buxton, K.E.; Scalf, M.; Jarrard, D.F.; Smith, L.M. HyPR-MS for Multiplexed Discovery of MALAT1, NEAT1, and NORAD lncRNA Protein Interactomes. J. Proteome Res. 2018, 17, 3022–3038. [Google Scholar] [CrossRef]
- Knoener, R.A.; Becker, J.T.; Scalf, M.; Sherer, N.M.; Smith, L.M. Elucidating the in vivo interactome of HIV-1 RNA by hybridization capture and mass spectrometry. Sci. Rep. 2017, 7, 16965. [Google Scholar] [CrossRef]
- Spiniello, M.; Steinbrink, M.I.; Cesnik, A.J.; Miller, R.M.; Scalf, M.; Shortreed, M.R.; Smith, L.M. Comprehensive in vivo identification of the c-Myc mRNA protein interactome using HyPR-MS. RNA 2019, 25, 1337–1352. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, A.; Dixit, U.; Manvar, D.; Chaturvedi, N.; Pandey, V.N. Affinity capture and identification of host cell factors associated with hepatitis C virus (+) strand subgenomic RNA. Mol. Cell. Proteom. 2013, 12, 1539–1552. [Google Scholar] [CrossRef] [Green Version]
- Lenarcic, E.M.; Landry, D.M.; Greco, T.M.; Cristea, I.M.; Thompson, S.R. Thiouracil cross-linking mass spectrometry: A cell-based method to identify host factors involved in viral amplification. J. Virol. 2013, 87, 8697–8712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, S.L.; Soderblom, E.J.; Bradrick, S.S.; Garcia-Blanco, M.A. Identification of Proteins Bound to Dengue Viral RNA In Vivo Reveals New Host Proteins Important for Virus Replication. MBio 2016, 7, e01865-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matia-Gonzalez, A.M.; Iadevaia, V.; Gerber, A.P. A versatile tandem RNA isolation procedure to capture in vivo formed mRNA-protein complexes. Methods 2017, 118, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogell, B.; Fischer, B.; Rettel, M.; Krijgsveld, J.; Castello, A.; Hentze, M.W. Specific RNP capture with antisense LNA/DNA mixmers. RNA 2017, 23, 1290–1302. [Google Scholar] [CrossRef] [Green Version]
- Theil, K.; Imami, K.; Rajewsky, N. Identification of proteins and miRNAs that specifically bind an mRNA in vivo. Nat. Commun. 2019, 10, 4205. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Spitale, R.C.; Chang, H.Y. Technologies to probe functions and mechanisms of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015, 22, 29–35. [Google Scholar] [CrossRef]
- Simon, M.D.; Wang, C.I.; Kharchenko, P.V.; West, J.A.; Chapman, B.A.; Alekseyenko, A.A.; Borowsky, M.L.; Kuroda, M.I.; Kingston, R.E. The genomic binding sites of a noncoding RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20497–20502. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef] [Green Version]
- Kazimierczyk, M.; Kasprowicz, M.K.; Kasprzyk, M.E.; Wrzesinski, J. Human Long Noncoding RNA Interactome: Detection, Characterization and Function. Int. J. Mol. Sci. 2020, 21, 1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, D.; Zhang, Z.; Chaubey, B.; Pandey, V.N. Identification of cellular factors associated with the 3’-nontranslated region of the hepatitis C virus genome. Mol. Cell. Proteom. 2006, 5, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Zeng, F.; Peritz, T.; Kannanayakal, T.J.; Kilk, K.; Eiriksdottir, E.; Langel, U.; Eberwine, J. A protocol for PAIR: PNA-assisted identification of RNA binding proteins in living cells. Nat. Protoc. 2006, 1, 920–927. [Google Scholar] [CrossRef]
- Phillips, S.L.; Garcia-Blanco, M.A.; Bradrick, S.S. Antisense-mediated affinity purification of dengue virus ribonucleoprotein complexes from infected cells. Methods 2015, 91, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.C.S.; Maurer-Stroh, S.; Wan, Y.; Sessions, O.M.; de Sessions, P.F. A novel method for the capture-based purification of whole viral native RNA genomes. AMB Express 2019, 9, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, T.; Smith, S.G.; Gaughan, K.; Oglesby, I.K.; O’Neill, S.; McElvaney, N.G.; Greene, C.M. Isolation and identification of cell-specific microRNAs targeting a messenger RNA using a biotinylated anti-sense oligonucleotide capture affinity technique. Nucleic Acids Res. 2013, 41, e71. [Google Scholar] [CrossRef] [Green Version]
- Iadevaia, V.; Matia-Gonzalez, A.M.; Gerber, A.P. An Oligonucleotide-based Tandem RNA Isolation Procedure to Recover Eukaryotic mRNA-Protein Complexes. J. Vis. Exp. 2018, 58223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Sun, W.; Shi, T.; Lu, P.; Zhuang, M.; Liu, J.L. Capturing RNA-protein interaction via CRUIS. Nucleic Acids Res. 2020, 48, e52. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Li, J.; Zhu, X.; Wang, X.; Fan, L.; Sun, W.; Liao, L.; Zhang, J.; Li, X.; Ye, J.; et al. CRISPR-assisted detection of RNA-protein interactions in living cells. Nat. Methods 2020, 17, 685–688. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Method | RNA Type | Target RNA | Crosslinker; ASO Probes | Ref. |
|---|---|---|---|---|
| Chart MS | lncRNA | NEAT1, MALAT1 | FA; 25-mer DNA, 3′TEG-biotin | [64] |
| ChIRP-MS | lncRNA | XIST | FA; 43 × 20-mer DNA, 3′TEG-biotin. | [65] |
| lncRNA | MALAT1 | FA; 32 × 20-mer DNA, 3′TEG-biotin | [66] | |
| RAP-MS | lncRNA | XIST | UV; 142 × 90-mer DNA, 5′-biotin | [67] |
| iDRiP | lncRNA | XIST | UV; 9 × 25-mer DNA, 3′TEG-biotin | [68] |
| HyPR-MS | lncRNA | MALAT1, NEAT1, NORAD | FA; 2–3 × 25–30-mer DNA, 8-nts toehold, 3′-biotin | [69] |
| virus | HIV | FA; 30-mer DNA, 8-nts toehold, 3′-biotin | [70] | |
| mRNA | c-myc | FA; 2 × 25-mer DNA, 8-nts toehold, 3′-biotin | [71] | |
| PAIR (like) | virus | HCV(-) | 15-mer biotinylated PNA oligo | [72] |
| TUX-MS | virus | Polio | UV; oligo(dT)25 beads | [73] |
| n.s. | virus | DENV | UV; 10 × 17–33-mer DNA, 5′-TEG-biotin | [74] |
| TRIP | mRNA | p27, gld-1, Pfk2 | UV; 21–25-mer 2′O-Met RNA, 3′-biotin | [75] |
| Specific RNA capture | mRNA rRNA | Rluc-sxl (in vitro) 18S | UV; 20-mer LNA with C6 amine-linker | [76] |
| vIPR | mRNA | gld-1::gfp, gfp::lin-41 | UV, FA; 10 × 20-mer DNA, 3′-TEG- biotin | [77] |
| mRNA | gld-1, lin-41, alg-1 | UV; 10 × 20-mer DNA, 3′-TEG- biotin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerber, A.P. RNA-Centric Approaches to Profile the RNA–Protein Interaction Landscape on Selected RNAs. Non-Coding RNA 2021, 7, 11. https://doi.org/10.3390/ncrna7010011
Gerber AP. RNA-Centric Approaches to Profile the RNA–Protein Interaction Landscape on Selected RNAs. Non-Coding RNA. 2021; 7(1):11. https://doi.org/10.3390/ncrna7010011
Chicago/Turabian StyleGerber, André P. 2021. "RNA-Centric Approaches to Profile the RNA–Protein Interaction Landscape on Selected RNAs" Non-Coding RNA 7, no. 1: 11. https://doi.org/10.3390/ncrna7010011
APA StyleGerber, A. P. (2021). RNA-Centric Approaches to Profile the RNA–Protein Interaction Landscape on Selected RNAs. Non-Coding RNA, 7(1), 11. https://doi.org/10.3390/ncrna7010011