
Nanoliposomal Delivery of MicroRNA-203 Suppresses Migration of Triple-Negative Breast Cancer through Distinct Target Suppression

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Overexpression of miR-203 through Nanoliposomal Encapsulation
2.2. Overexpression and Delivery of miR-203 Suppress Migration
2.3. Delivery of miR-203 Suppresses Sphere Formation in HMLE Twist Cells
2.4. Overexpression and Delivery of miR-203 Alters Expression of Target mRNA
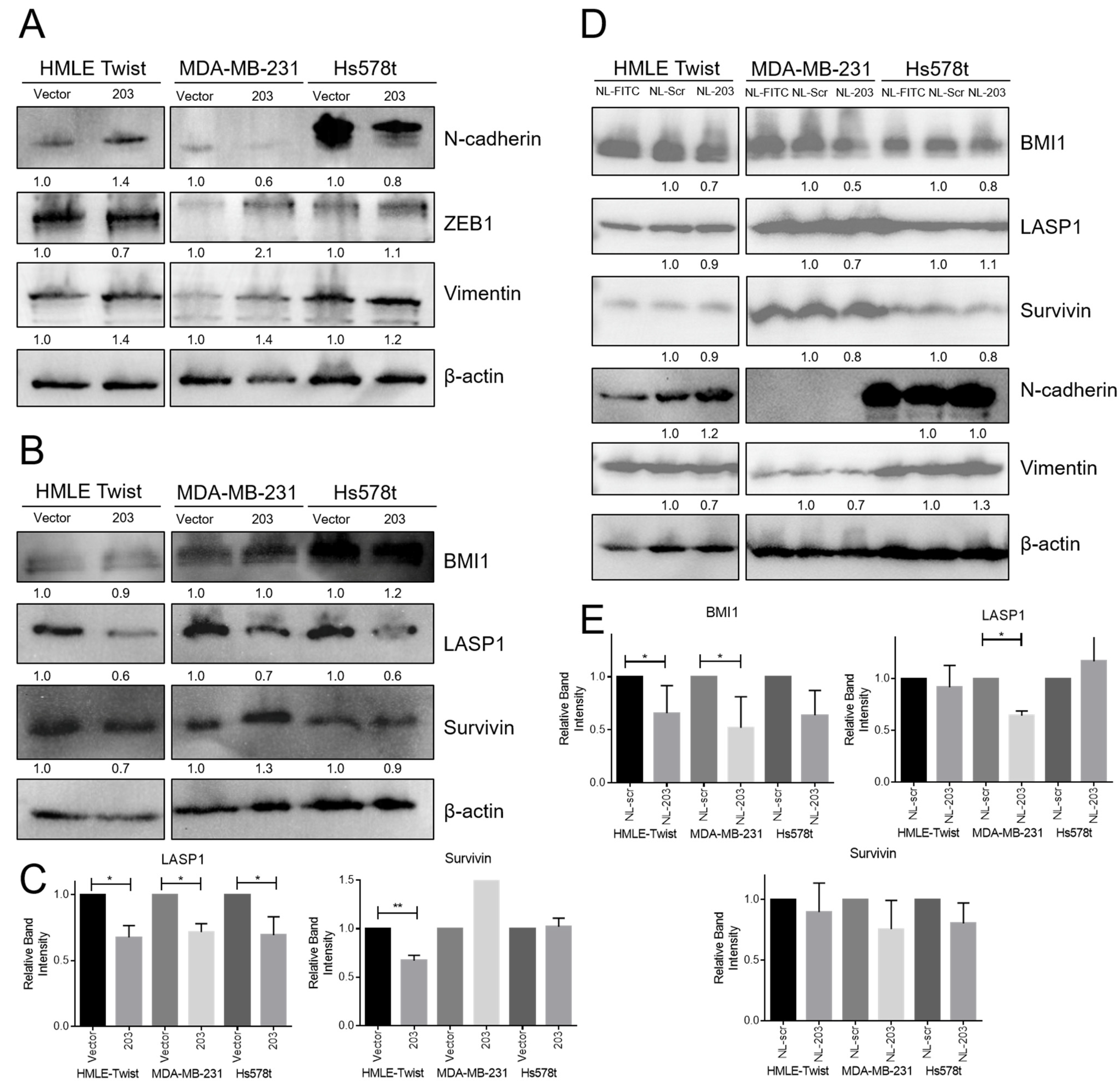
2.5. Overexpression of miR-203 Suppresses LASP1 and Survivin (BIRC5) Protein Expression, and Delivery of miR-203 Suppresses BMI1 Protein Expression
2.6. Loss of MIR203 Copy Number in TNBC
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Viral Transduction
4.3. Nanoliposome Synthesis
4.4. RNA Isolation and Quantitative Reverse-Transcription PCR
4.5. Nanostring Analysis
4.6. Immunoblotting
4.7. Cell Migration Assay
4.8. Mammosphere Assay
4.9. Patient Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Breast Cancer Facts & Figures; The Society: Atlanta, GA, USA, 2021. [Google Scholar]
- Pareja, F.; Geyer, F.C.; Marchiò, C.; Burke, K.A.; Weigelt, B.; Reis-Filho, J.S. Triple-negative breast cancer: The importance of molecular and histologic subtyping, and recognition of low-grade variants. NPJ Breast Cancer 2016, 2, 16036. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Lee, D.K.; Feng, Z.; Xu, Y.; Bu, W.; Li, Y.; Liao, L.; Xu, J. Breast tumor cell-specific knockout of Twist1 inhibits cancer cell plasticity, dissemination, and lung metastasis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 11494–11499. [Google Scholar] [CrossRef] [Green Version]
- Revenco, T.; Nicodeme, A.; Pastushenko, I.; Sznurkowska, M.K.; Latil, M.; Sotiropoulou, P.A.; Dubois, C.; Moers, V.; Lemaire, S.; de Maertelaer, V.; et al. Context Dependency of Epithelial-to-Mesenchymal Transition for Metastasis. Cell Rep. 2019, 29, 1458–1468.e1453. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Mayor, R. Tuning collective cell migration by cell-cell junction regulation. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Herranz, N.; Pasini, D.; Diaz, V.M.; Franci, C.; Gutierrez, A.; Dave, N.; Escriva, M.; Hernandez-Munoz, I.; Di Croce, L.; Helin, K.; et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol. Cell Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Werden, S.J.; Sphyris, N.; Sarkar, T.R.; Paranjape, A.N.; LaBaff, A.M.; Taube, J.H.; Hollier, B.G.; Ramirez-Pena, E.Q.; Soundararajan, R.; den Hollander, P.; et al. Phosphorylation of serine 367 of FOXC2 by p38 regulates ZEB1 and breast cancer metastasis, without impacting primary tumor growth. Oncogene 2016, 35, 5977–5988. [Google Scholar] [CrossRef] [PubMed]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Dutta, A. MicroRNAs in cancer. Annu. Rev. Pathol. 2009, 4, 199–227. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef] [Green Version]
- Title, A.C.; Hong, S.-J.; Pires, N.D.; Hasenöhrl, L.; Godbersen, S.; Stokar-Regenscheit, N.; Bartel, D.P.; Stoffel, M. Genetic dissection of the miR-200-Zeb1 axis reveals its importance in tumor differentiation and invasion. Nat. Commun. 2018, 9, 4671. [Google Scholar] [CrossRef]
- Zaravinos, A. The Regulatory Role of MicroRNAs in EMT and Cancer. J. Oncol. 2015, 2015, 865816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chim, C.S.; Wong, K.Y.; Leung, C.Y.; Chung, L.P.; Hui, P.K.; Chan, S.Y.; Yu, L. Epigenetic inactivation of the hsa-miR-203 in haematological malignancies. J. Cell Mol. Med. 2011, 15, 2760–2767. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.; Bai, Y.; Qiu, S.; Zheng, L.; Huang, L.; Liu, T.; Wang, X.; Liu, Y.; Xu, N.; Yan, X.; et al. MiR-203 downregulation is responsible for chemoresistance in human glioblastoma by promoting epithelial-mesenchymal transition via SNAI2. Oncotarget 2015, 6, 8914–8928. [Google Scholar] [CrossRef] [PubMed]
- Diao, Y.; Guo, X.; Jiang, L.; Wang, G.; Zhang, C.; Wan, J.; Jin, Y.; Wu, Z. miR-203, a tumor suppressor frequently down-regulated by promoter hypermethylation in rhabdomyosarcoma. J. Biol. Chem. 2014, 289, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Saini, S.; Majid, S.; Yamamura, S.; Tabatabai, L.; Suh, S.O.; Shahryari, V.; Chen, Y.; Deng, G.; Tanaka, Y.; Dahiya, R. Regulatory Role of mir-203 in Prostate Cancer Progression and Metastasis. Clin. Cancer Res. 2011, 17, 5287–5298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benaich, N.; Woodhouse, S.; Goldie, S.J.; Mishra, A.; Quist, S.R.; Watt, F.M. Rewiring of an Epithelial Differentiation Factor, miR-203, to Inhibit Human Squamous Cell Carcinoma Metastasis. Cell Rep. 2014, 9, 104–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.-T.; Mock, K.; Ruh, M.; Schüler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef]
- Bueno, M.J.; Perez de Castro, I.; Gomez de Cedron, M.; Santos, J.; Calin, G.A.; Cigudosa, J.C.; Croce, C.M.; Fernandez-Piqueras, J.; Malumbres, M. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell 2008, 13, 496–506. [Google Scholar] [CrossRef] [Green Version]
- Taube, J.H.; Malouf, G.G.; Lu, E.; Sphyris, N.; Vijay, V.; Ramachandran, P.P.; Ueno, K.R.; Gaur, S.; Nicoloso, M.S.; Rossi, S.; et al. Epigenetic silencing of microRNA-203 is required for EMT and cancer stem cell properties. Sci. Rep. 2013, 3, 2687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Li, G.Y.; Jiang, L.; Jia, L.; Zhang, Z.; Li, X.; Wang, R.; Zhou, M.; Zhou, Y.; Zeng, Z.; et al. Long noncoding RNA CAR10 promotes lung adenocarcinoma metastasis via miR-203/30/SNAI axis. Oncogene 2019, 38, 3061–3076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.; Garikapati, K.R.; Makani, V.K.K.; Nair, A.D.; Vangara, N.; Bhadra, U.; Bhadra, M.P. Regulating BMI1 expression via miRNAs promote Mesenchymal to Epithelial Transition (MET) and sensitizes breast cancer cell to chemotherapeutic drug. PLoS ONE 2018, 13, e0190245. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.F.; Xu, C.; Chen, J.; Ding, C.; Xu, Z.L.; Li, C.; Zhao, J. MicroRNA-203 inhibits cellular proliferation and invasion by targeting Bmi1 in non-small cell lung cancer. Oncol. Lett. 2015, 9, 2639–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Ungvári, I.; Hadadi, E.; Virág, V.; Bikov, A.; Nagy, A.; Semsei, A.F.; Gálffy, G.; Tamási, L.; Horváth, I.; Szalai, C. Implication of BIRC5 in asthma pathogenesis. Int. Immunol. 2012, 24, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zheng, X.; Shen, C.; Shi, Y. MicroRNA-203 suppresses cell proliferation and migration by targeting BIRC5 and LASP1 in human triple-negative breast cancer cells. J. Exp. Clin. Cancer Res. 2012, 31. [Google Scholar] [CrossRef] [Green Version]
- Hailer, A.; Grunewald, T.G.P.; Orth, M.; Reiss, C.; Kneitz, B.; Spahn, M.; Butt, E. Loss of tumor suppressor mir-203 mediates overexpression of LIM and SH3 Protein 1 (LASP1) in high-risk prostate cancer thereby increasing cell proliferation and migration. Oncotarget 2014, 5, 4144–4153. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wan, G.; Spizzo, R.; Ivan, C.; Mathur, R.; Hu, X.; Ye, X.; Lu, J.; Fan, F.; Xia, L.; et al. miR-203 induces oxaliplatin resistance in colorectal cancer cells by negatively regulating ATM kinase. Mol. Oncol. 2014, 8, 83–92. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, J.; Xiao, M.; Yang, J.; Zhang, N. miR-203 Suppresses Bladder Cancer Cell Growth and Targets Twist1. Oncol. Res. 2018, 26, 1155–1165. [Google Scholar] [CrossRef]
- He, S.; Zhang, G.; Dong, H.; Ma, M.; Sun, Q. miR-203 facilitates tumor growth and metastasis by targeting fibroblast growth factor 2 in breast cancer. OncoTargets Ther. 2016, 9, 6203–6210. [Google Scholar] [CrossRef] [Green Version]
- Mine, M.; Yamaguchi, K.; Sugiura, T.; Chigita, S.; Yoshihama, N.; Yoshihama, R.; Hiyake, N.; Kobayashi, Y.; Mori, Y. miR-203 Inhibits Frizzled-2 Expression via CD82/KAI1 Expression in Human Lung Carcinoma Cells. PLoS ONE 2015, 10, e0131350. [Google Scholar]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Wang, J.; Wu, X.; Shen, P.; Wang, J.; Shen, Y.; Shen, Y.; Webster, T.J.; Deng, J. Applications of Inorganic Nanomaterials in Photothermal Therapy Based on Combinational Cancer Treatment. Int. J. Nanomed. 2020, 15, 1903–1914. [Google Scholar] [CrossRef] [Green Version]
- Malam, Y.; Loizidou, M.; Seifalian, A.M. Liposomes and nanoparticles: Nanosized vehicles for drug delivery in cancer. Trends Pharmacol. Sci. 2009, 30, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Etheridge, M.L.; Campbell, S.A.; Erdman, A.G.; Haynes, C.L.; Wolf, S.M.; McCullough, J. The big picture on nanomedicine: The state of investigational and approved nanomedicine products. Nanomedicine 2013, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar] [CrossRef] [Green Version]
- Lujan, H.; Griffin, W.C.; Taube, J.H.; Sayes, C.M. Synthesis and characterization of nanometer-sized liposomes for encapsulation and microRNA transfer to breast cancer cells. Int. J. Nanomed. 2019, 14, 5159–5173. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.I.; Yeh, M.K. Clinical development of liposome-based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomed. 2012, 7, 49–60. [Google Scholar]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Chen, Y.; Zhao, J.; Kong, F.; Zhang, Y. miR-203 reverses chemoresistance in p53-mutated colon cancer cells through downregulation of Akt2 expression. Cancer Lett. 2011, 304, 52–59. [Google Scholar] [CrossRef]
- To, K.K.; Leung, W.W.; Ng, S.S. A novel miR-203-DNMT3b-ABCG2 regulatory pathway predisposing colorectal cancer development. Mol. Carcinog. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.W.; Liu, G.P.; Wang, K.Z. miR-203 acts as a tumor suppressor gene in osteosarcoma by regulating RAB22A. PLoS ONE 2015, 10, e0132225. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Tao, F.; Zhang, B.; Dong, J.-T.; Zhang, Z. The miR-203/SNAI2 axis regulates prostate tumor growth, migration, angiogenesis and stemness potentially by modulating GSK-3β/β-CATENIN signal pathway. IUBMB Life 2018, 70, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhang, B.; Li, W.; Fu, L.; Fu, L.; Zhu, Z.; Dong, J.T. Epigenetic Silencing of miR-203 Upregulates SNAI2 and Contributes to the Invasiveness of Malignant Breast Cancer Cells. Genes Cancer 2011, 2, 782–791. [Google Scholar] [CrossRef]
- Ru, P.; Steele, R.; Hsueh, E.C.; Ray, R.B. Anti-mir-203 upregulates socs3 expression in breast cancer cells and enhances cisplatin chemosensitivity. Genes Cancer 2011, 2, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Kim, J.S.; Park, S.Y.; Kim, H.J.; Yu, S.K.; Kim, C.S.; Chun, H.S.; Kim, J.; Park, J.T.; Go, D.; et al. miR-203 downregulates Yes-1 and suppresses oncogenic activity in human oral cancer cells. J. Biosci. Bioeng. 2015, 120, 351–358. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.F.R.; Fulton, R.; McLellan, M.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.; Fulton, L.; Dooling, D.; Ding, L.; Mardis, E.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repecka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.H.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Thorsen, S.B.; Obad, S.; Jensen, N.F.; Stenvang, J.; Kauppinen, S. The therapeutic potential of microRNAs in cancer. Cancer J. 2012, 18, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, Y.; Peng, H.; Chen, Y.; Zhu, P.; Huang, Y. Recent progress in microRNA delivery for cancer therapy by non-viral synthetic vectors. Adv. Drug Deliv. Rev. 2015, 81, 142–160. [Google Scholar] [CrossRef]
- Pramanik, D.; Campbell, N.R.; Karikari, C.; Chivukula, R.; Kent, O.A.; Mendell, J.T.; Maitra, A. Restitution of tumor suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol. Cancer Ther. 2011, 10, 1470–1480. [Google Scholar] [CrossRef] [Green Version]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [Green Version]
- Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Patrawala, L.; Brown, D.; Bader, A.G. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010, 70, 5923–5930. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, M.; Jung, E.J.; Shah, M.Y.; Lu, C.; Spizzo, R.; Shimizu, M.; Han, H.D.; Ivan, C.; Rossi, S.; Zhang, X.; et al. Therapeutic synergy between microRNA and siRNA in ovarian cancer treatment. Cancer Discov. 2013, 3, 1302–1315. [Google Scholar] [CrossRef] [Green Version]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zhou, Y.; Wang, C.; Zhang, S.; Pan, Y.; Wang, Y.; Yan, X.; Zhang, J.; Zhang, C.Y.; et al. miR-203 suppresses the proliferation and migration and promotes the apoptosis of lung cancer cells by targeting SRC. PLoS ONE 2014, 9, e105570. [Google Scholar] [CrossRef] [PubMed]
- Orom, U.A.; Lim, M.K.; Savage, J.E.; Jin, L.; Saleh, A.D.; Lisanti, M.P.; Simone, N.L. MicroRNA-203 regulates caveolin-1 in breast tissue during caloric restriction. Cell Cycle 2012, 11, 1291–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.W.; Kuo, C.T.; Chen, J.H.; Goodfellow, P.J.; Huang, T.H.; Rader, J.S.; Uyar, D.S. Hypermethylation of miR-203 in endometrial carcinomas. Gynecol. Oncol. 2014, 133, 340–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.H.; Hsu, D.S.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Paranjape, A.N.; Balaji, S.A.; Mandal, T.; Krushik, E.V.; Nagaraj, P.; Mukherjee, G.; Rangarajan, A. Bmi1 regulates self-renewal and epithelial to mesenchymal transition in breast cancer cells through Nanog. BMC Cancer 2014, 14, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.; Johnson, K.S.; Lujan, H.; Pradhan, S.H.; Sayes, C.M.; Taube, J.H. Nanoliposomal Delivery of MicroRNA-203 Suppresses Migration of Triple-Negative Breast Cancer through Distinct Target Suppression. Non-Coding RNA 2021, 7, 45. https://doi.org/10.3390/ncrna7030045
Song S, Johnson KS, Lujan H, Pradhan SH, Sayes CM, Taube JH. Nanoliposomal Delivery of MicroRNA-203 Suppresses Migration of Triple-Negative Breast Cancer through Distinct Target Suppression. Non-Coding RNA. 2021; 7(3):45. https://doi.org/10.3390/ncrna7030045
Chicago/Turabian StyleSong, Shuxuan, Kelsey S. Johnson, Henry Lujan, Sahar H. Pradhan, Christie M. Sayes, and Joseph H. Taube. 2021. "Nanoliposomal Delivery of MicroRNA-203 Suppresses Migration of Triple-Negative Breast Cancer through Distinct Target Suppression" Non-Coding RNA 7, no. 3: 45. https://doi.org/10.3390/ncrna7030045
APA StyleSong, S., Johnson, K. S., Lujan, H., Pradhan, S. H., Sayes, C. M., & Taube, J. H. (2021). Nanoliposomal Delivery of MicroRNA-203 Suppresses Migration of Triple-Negative Breast Cancer through Distinct Target Suppression. Non-Coding RNA, 7(3), 45. https://doi.org/10.3390/ncrna7030045