
tRNA Synthetases Are Recruited to Yeast Ribosomes by rRNA Expansion Segment 7L but Do Not Require Association for Functionality


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Yeast Strains and Cultivation
2.2. Polysomes
2.3. Western Blot
2.4. Metabolic Labeling
2.5. Total RNA Isolation
2.6. Poisoned Primer Extension
2.7. tRNA Northern Blots
2.8. rRNA Northern Blot
3. Results
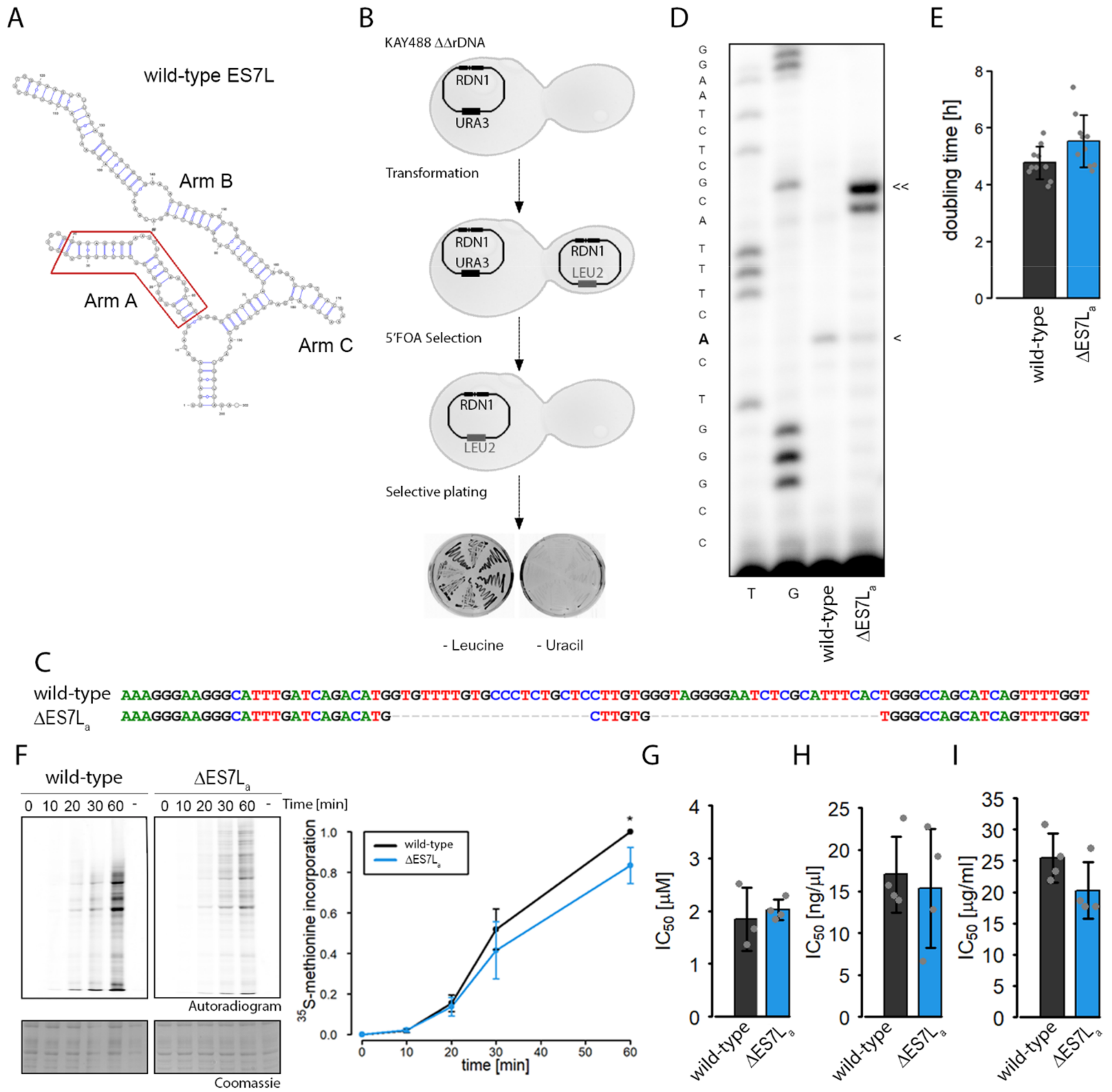
3.1. Removal of Expansion Segment ES7La Slightly Reduces Cellular Fitness and Translation Capacity
3.2. ES7La Deletion Redistributes Aminoacyl-tRNA Synthetases from Ribosomes
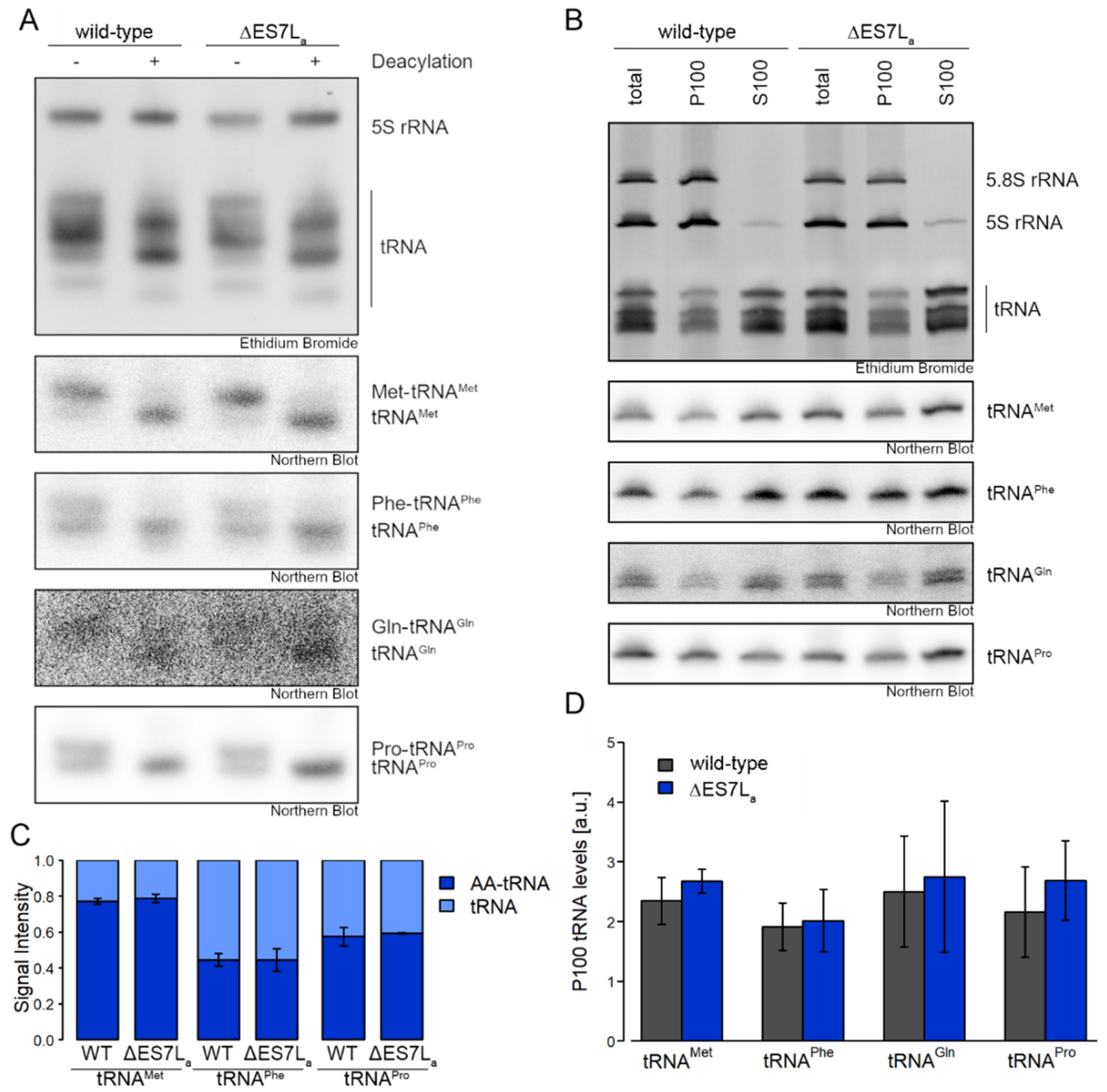
3.3. Aminoacyl-tRNA Synthetases Maintain Activity when Dissociated from the Ribosome
4. Discussion
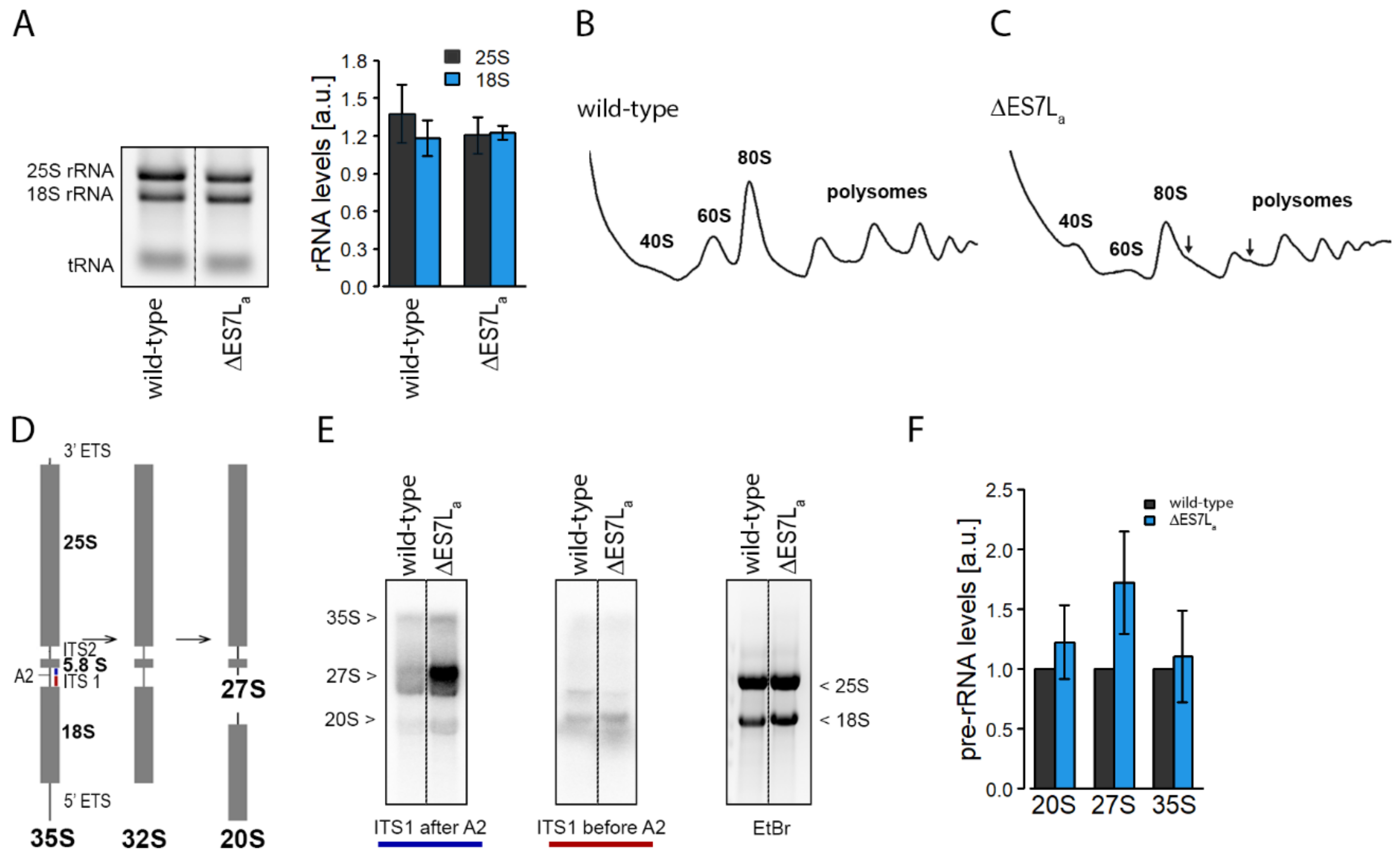
4.1. ∆ES7La rRNA Maturation Is Perturbed and Translation Efficiency Reduced
4.2. Aminoacyl-tRNA Synthetase Recruitment Is No Prerequisite for tRNA Charging
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fox, G.E. Origin and Evolution of the Ribosome. Cold Spring Harb. Perspect. Biol. 2010, 2, a003483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnikov, S.; Ben-Shem, A.; Garreau De Loubresse, N.; Jenner, L.; Yusupova, G.; Yusupov, M. One Core, Two Shells: Bacterial and Eukaryotic Ribosomes. Nat. Struct. Mol. Biol. 2012, 19, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.S.; Bernier, C.R.; Hsiao, C.; Norris, A.M.; Kovacs, N.A.; Waterbury, C.C.; Stepanov, V.G.; Harvey, S.C.; Fox, G.E.; Wartell, R.M.; et al. Evolution of the Ribosome at Atomic Resolution. Proc. Natl. Acad. Sci. USA 2014, 111, 10251–10256. [Google Scholar] [CrossRef] [Green Version]
- Petrov, A.S.; Gulen, B.; Norris, A.M.; Kovacs, N.A.; Bernier, C.R.; Lanier, K.A.; Fox, G.E.; Harvey, S.C.; Wartell, R.M.; Hud, N.V.; et al. History of the Ribosome and the Origin of Translation. Proc. Natl. Acad. Sci. USA 2015, 112, 15396–15401. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, M.; Woolford, J.L., Jr. Eukaryote-Specific RRNA Expansion Segments Function in Ribosome Biogenesis. RNA 2016, 22, 1153–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Shem, A.; Garreau de Loubresse, N.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The Structure of the Eukaryotic Ribosome at 3.0 A Resolution. Science 2011, 334, 1524–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, V.; Rauscher, R.; Reuther, J.; Gharib, W.H.; Koch, M.; Polacek, N. RRNA Expansion Segment 27Lb Modulates the Factor Recruitment Capacity of the Yeast Ribosome and Shapes the Proteome. Nucleic Acids Res. 2020, 48, 3244–3256. [Google Scholar] [CrossRef]
- Fujii, K.; Susanto, T.T.; Saurabh, S.; Barna, M. Decoding the Function of Expansion Segments in Ribosomes. Mol. Cell 2018, 72, 1013–1020. [Google Scholar] [CrossRef] [Green Version]
- Leppek, K.; Fujii, K.; Quade, N.; Susanto, T.T.; Boehringer, D.; Lenarčič, T.; Xue, S.; Genuth, N.R.; Ban, N.; Barna, M. Gene- and Species-Specific Hox MRNA Translation by Ribosome Expansion Segments. Mol. Cell 2020, 80, 980–995. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Williams, L.D.; Pestov, D.G.; Shcherbik, N. Proteotoxic Stress Promotes Entrapment of Ribosomes and Misfolded Proteins in a Shared Cytosolic Compartment. Nucleic Acids Res. 2021, 48, 3888–3905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinskie, J.A.; Ghosh, A.; Trainor, B.M.; Shedlovskiy, D.; Pestov, D.G.; Shcherbik, N. Iron-Dependent Cleavage of Ribosomal RNA during Oxidative Stress in the Yeast Saccharomyces Cerevisiae. J. Biol. Chem. 2018, 293, 14237–14248. [Google Scholar] [CrossRef] [Green Version]
- Shedlovskiy, D.; Zinskie, J.A.; Gardner, E.; Pestov, D.G.; Shcherbik, N. Endonucleolytic Cleavage in the Expansion Segment 7 of 25S RRNA Is an Early Marker of Low-Level Oxidative Stress in Yeast. J. Biol. Chem. 2017, 292, 18469–18485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez Ramos, L.M.; Smeekens, J.M.; Kovacs, N.A.; Bowman, J.C.; Wartell, R.M.; Wu, R.; Williams, L.D. Yeast RRNA Expansion Segments: Folding and Function. J. Mol. Biol. 2016, 428, 4048–4059. [Google Scholar] [CrossRef] [Green Version]
- Negrutskii, B.S.; Deutscher, M.P. A Sequestered Pool of Aminoacyl-TRNA in Mammalian Cells. Proc. Natl. Acad. Sci. USA 1992, 89, 3601–3604. [Google Scholar] [CrossRef] [Green Version]
- Negrutskii, B.S.; Deutscher, M.P. Channeling of Aminoacyl-TRNA for Protein Synthesis in Vivo. Proc. Natl. Acad. Sci. USA 1991, 88, 4991–4995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Fedyunin, I.; Miekley, O.; Valleriani, A.; Moura, A.; Ignatova, Z. Global and Local Depletion of Ternary Complex Limits Translational Elongation. Nucleic Acids Res. 2010, 38, 4778–4787. [Google Scholar] [CrossRef] [Green Version]
- Raina, M.; Elgamal, S.; Santangelo, T.J.; Ibba, M. Association of a Multi-Synthetase Complex with Translating Ribosomes in the Archaeon Thermococcus Kodakarensis. FEBS Lett. 2012, 586, 2232–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godinic-Mikulcic, V.; Jaric, J.; Greber, B.J.; Franke, V.; Hodnik, V.; Anderluh, G.; Ban, N.; Weygand-Durasevic, I. Archaeal Aminoacyl-TRNA Synthetases Interact with the Ribosome to Recycle TRNAs. Nucleic Acids Res. 2014, 42, 5191–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laporte, D.; Huot, J.L.; Bader, G.; Enkler, L.; Senger, B.; Becker, H.D. Exploring the Evolutionary Diversity and Assembly Modes of Multi-Aminoacyl-TRNA Synthetase Complexes: Lessons from Unicellular Organisms. FEBS Lett. 2014, 588, 4268–4278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, A.; Netzer, N.; Strader, M.B.; Das, S.R.; Chen, C.Y.; Gibbs, J.; Pierre, P.; Bennink, J.R.; Yewdell, J.W. RNA Binding Targets Aminoacyl-TRNA Synthetases to Translating Ribosomes. J. Biol. Chem. 2011, 286, 20688–20700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminska, M.; Havrylenko, S.; Decottignies, P.; le Maréchal, P.; Negrutskii, B.; Mirande, M. Dynamic Organization of Aminoacyl-TRNA Synthetase Complexes in the Cytoplasm of Human Cells. J. Biol. Chem. 2009, 284, 13746–13754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, A.; Enger, M.D. Subcellular Distribution of Aminoacyl-Transfer RNA Synthetases in Chinese Hamster Ovary Cell Culture. J. Mol. Biol. 1973, 79, 285–293. [Google Scholar] [CrossRef]
- Simos, G.; Segref, A.; Fasiolo, F.; Hellmuth, K.; Shevchenko, A.; Mann, M.; Hurt, E.C. The Yeast Protein Arc1p Binds to TRNA and Functions as a Cofactor for the Methionyl- and Glutamyl-TRNA Synthetases. EMBO J. 1996, 15, 5437–5448. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Kapur, M.; Diedrich, J.K.; Yates, J.R.; Ackerman, S.L.; Schimmel, P. Regulation of Ex-Translational Activities Is the Primary Function of the Multi-TRNA Synthetase Complex. Nucleic Acids Res. 2021, 49, 3603–3616. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Nilsen, T.W. Poisoned Primer Extension. Cold Spring Harb. Protoc. 2015, 2015. [Google Scholar] [CrossRef]
- Armache, J.P.; Jarasch, A.; Anger, A.M.; Villa, E.; Becker, T.; Bhushan, S.; Jossinet, F.; Habeck, M.; Dindar, G.; Franckenberg, S.; et al. Cryo-EM Structure and RRNA Model of a Translating Eukaryotic 80S Ribosome at 5.5-A Resolution. Proc. Natl. Acad. Sci. USA 2010, 107, 19748–19753. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, N.; Singh, C.R.; Udagawa, T.; Wang, S.; Thorson, E.; Winter, Z.; Ohira, T.; Ii, M.; Valášek, L.; Brown, S.J.; et al. Yeast 18S RRNA Is Directly Involved in the Ribosomal Response to Stringent AUG Selection during Translation Initiation. J. Biol. Chem. 2010, 285, 32200–32212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuker, M. Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Baierlein, C.; Hackmann, A.; Gross, T.; Henker, L.; Hinz, F.; Krebber, H. Monosome Formation during Translation Initiation Requires the Serine/Arginine-Rich Protein Npl3. Mol. Cell. Biol. 2013, 33, 4811–4823. [Google Scholar] [CrossRef] [Green Version]
- Behrmann, E.; Loerke, J.; Budkevich, T.V.; Yamamoto, K.; Schmidt, A.; Penczek, P.A.; Vos, M.R.; Bürger, J.; Mielke, T.; Scheerer, P.; et al. Structural Snapshots of Actively Translating Human Ribosomes. Cell 2015, 161, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Jeeninga, R.E.; van Delft, Y.; de Graaff-Vincent, M.; Dirks-Mulder, A.; Venema, J.; Raue, H.A. Variable Regions V13 and V3 of Saccharomyces Cerevisiae Contain Structural Features Essential for Normal Biogenesis and Stability of 5.8S and 25S RRNA. RNA 1997, 3, 476–488. [Google Scholar] [PubMed]
- Gamalinda, M.; Woolford, J.L. Deletion of L4 Domains Reveals Insights into the Importance of Ribosomal Protein Extensions in Eukaryotic Ribosome Assembly. RNA 2014, 20, 1725–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- USSERY, M.A.; TANAKA, W.K.; HARDESTY, B. Subcellular Distribution of Aminoacyl-TRNA Synthetases in Various Eukaryotic Cells. Eur. J. Biochem. 1977, 72, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Ogata, K.; Ohno, R.; Morioka, S.; Terao, K. Further Study on Association of 5SrRNA-L5 Protein Complex and Methionyl-TRNA to Methionyl-TRNA Synthetase in the Macromolecular Aminoacyl-TRNA Synthetase Complex. J. Biochem. 1996, 120, 869–880. [Google Scholar] [CrossRef]
- Tscherne, J.S.; Weinstein, B.; Lanks, K.W.; Gersten, N.B.; Cantor, C.R. Phenylalanyl Transfer Ribonucleic Acid Synthetase Activity Associated with Rat Liver Ribosomes and Microsomes. Biochemistry 2002, 12, 3859–3865. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, C.D.; Prætorius-Ibba, M.; Ibba, M. An Aminoacyl-TRNA Synthetase:Elongation Factor Complex for Substrate Channeling in Archaeal Translation. Nucleic Acids Res. 2007, 35, 6094–6102. [Google Scholar] [CrossRef] [Green Version]
- Locati, M.D.; Pagano, J.F.B.; Girard, G.; Ensink, W.A.; van Olst, M.; van Leeuwen, S.; Nehrdich, U.; Spaink, H.P.; Rauwerda, H.; Jonker, M.J.; et al. Expression of Distinct Maternal and Somatic 5.8S, 18S, and 28S RRNA Types during Zebrafish Development. RNA 2017, 23, 1188–1199. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Fujii, K.; Kovary, K.M.; Genuth, N.R.; Röst, H.L.; Teruel, M.N.; Barna, M. Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of MRNAs Genome-Wide. Mol. Cell 2017, 67, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simsek, D.; Tiu, G.C.; Flynn, R.A.; Byeon, G.W.; Leppek, K.; Xu, A.F.; Chang, H.Y.; Barna, M. The Mammalian Ribo-Interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell 2017, 169, 1051–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genuth, N.R.; Barna, M. Heterogeneity and Specialized Functions of Translation Machinery: From Genes to Organisms. Nat. Rev. Genet. 2018, 19, 431–452. [Google Scholar] [CrossRef] [PubMed]
- Rauscher, R.; Ignatova, Z. Timing during Translation Matters: Synonymous Mutations in Human Pathologies Influence Protein Folding and Function. Biochem. Soc. Trans. 2018, 46, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krauer, N.; Rauscher, R.; Polacek, N. tRNA Synthetases Are Recruited to Yeast Ribosomes by rRNA Expansion Segment 7L but Do Not Require Association for Functionality. Non-Coding RNA 2021, 7, 73. https://doi.org/10.3390/ncrna7040073
Krauer N, Rauscher R, Polacek N. tRNA Synthetases Are Recruited to Yeast Ribosomes by rRNA Expansion Segment 7L but Do Not Require Association for Functionality. Non-Coding RNA. 2021; 7(4):73. https://doi.org/10.3390/ncrna7040073
Chicago/Turabian StyleKrauer, Nina, Robert Rauscher, and Norbert Polacek. 2021. "tRNA Synthetases Are Recruited to Yeast Ribosomes by rRNA Expansion Segment 7L but Do Not Require Association for Functionality" Non-Coding RNA 7, no. 4: 73. https://doi.org/10.3390/ncrna7040073
APA StyleKrauer, N., Rauscher, R., & Polacek, N. (2021). tRNA Synthetases Are Recruited to Yeast Ribosomes by rRNA Expansion Segment 7L but Do Not Require Association for Functionality. Non-Coding RNA, 7(4), 73. https://doi.org/10.3390/ncrna7040073