
TGF-β-Upregulated Lnc-Nr6a1 Acts as a Reservoir of miR-181 and Mediates Assembly of a Glycolytic Complex
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
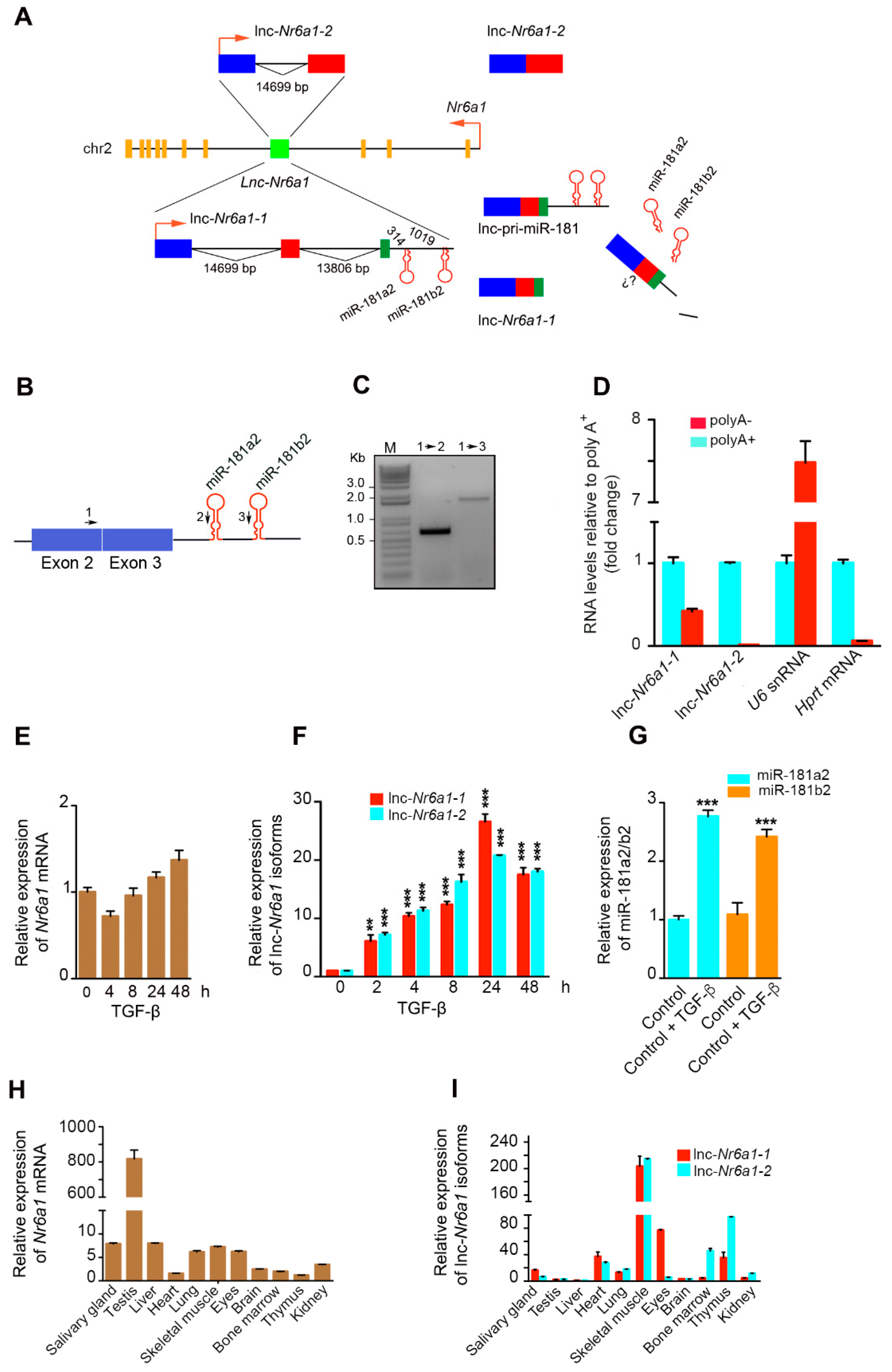
2.1. Upregulation of Lnc-Nr6a1 and Embedded miR-181a2 and miR-181b2 during EMT
2.2. Lnc-Nr6a1 Depletion Reduces Cell Adhesion and Alters Gene Expression
2.3. Lnc-Nr6a1 Protein Interactome
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Lentiviral Production and Infection Assay
4.4. Western Blot Analysis
4.5. Proliferation, Migration and Invasion Assays
4.6. Immunofluorescence Microscopy
4.7. RNA Isolation and RT-qPCR
4.8. Poly(A)+ and Poly(A)− RNA Separation
4.9. In Vitro Poly(A) Tailing and RACE
4.10. RNA Isolation from Mouse Tissues
4.11. Cell Fractionation
4.12. RNA-Seq Analysis
4.13. ChIP Sequencing
4.14. RNA Immunoprecipitation
4.15. CHIRP
4.16. CRISPR-SAM-Mediated Activation of Lnc-Nr6a1
4.17. CRISPR/Cas9-Mediated Deletion of Lnc-Nr6a1
4.18. iDRIP
4.19. Protein Identification Using Mass Spectrometry
4.20. Adhesion Assays
4.21. Anoikis Induction and Flow Cytometry
4.22. Seahorse Assay
4.23. Analysis of Tumor Data Sets
4.24. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Cheng, A.W.; Flytzanis, N.C.; Balsamo, M.; Condeelis, J.S.; Oktay, M.H.; Burge, C.B.; Gertler, F.B. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011, 7, e1002218. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Mateo, C.; Torres, B.; Gutierrez, G.; Pintor-Toro, J.A. Downregulation of Lnc-Spry1 mediates TGF-beta-induced epithelial-mesenchymal transition by transcriptional and posttranscriptional regulatory mechanisms. Cell Death Differ. 2017, 24, 785–797. [Google Scholar] [CrossRef]
- Zaravinos, A. The Regulatory Role of MicroRNAs in EMT and Cancer. J. Oncol. 2015, 2015, 865816. [Google Scholar] [CrossRef]
- Uszczynska-Ratajczak, B.; Lagarde, J.; Frankish, A.; Guigo, R.; Johnson, R. Towards a complete map of the human long non-coding RNA transcriptome. Nat. Rev. Genet. 2018, 19, 535–548. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Keniry, A.; Oxley, D.; Monnier, P.; Kyba, M.; Dandolo, L.; Smits, G.; Reik, W. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat. Cell Biol. 2012, 14, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhao, X.; Liu, Q.; Li, C.; Graves-Deal, R.; Cao, Z.; Singh, B.; Franklin, J.L.; Wang, J.; Hu, H.; et al. lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/beta-catenin signaling. Nat. Med. 2017, 23, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.Y.; Moriarity, B.S.; Gong, W.; Akiyama, R.; Tiwari, A.; Kawakami, H.; Ronning, P.; Reuland, B.; Guenther, K.; Beadnell, T.C.; et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 2014, 512, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hebert, M.C.; Zhang, Y.E. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef]
- Guerrero-Martinez, J.A.; Ceballos-Chavez, M.; Koehler, F.; Peiro, S.; Reyes, J.C. TGFbeta promotes widespread enhancer chromatin opening and operates on genomic regulatory domains. Nat. Commun. 2020, 11, 6196. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef]
- Minajigi, A.; Froberg, J.; Wei, C.; Sunwoo, H.; Kesner, B.; Colognori, D.; Lessing, D.; Payer, B.; Boukhali, M.; Haas, W.; et al. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 2015, 349, 6245. [Google Scholar] [CrossRef]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef]
- Nguyen, H.Q.; Bosco, G. Gene Positioning Effects on Expression in Eukaryotes. Annu Rev. Genet. 2015, 49, 627–646. [Google Scholar] [CrossRef]
- Dhir, A.; Dhir, S.; Proudfoot, N.J.; Jopling, C.L. Microprocessor mediates transcriptional termination of long noncoding RNA transcripts hosting microRNAs. Nat. Struct. Mol. Biol. 2015, 22, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Song, Y.J.; Prasanth, K.V. One locus with two roles: microRNA-independent functions of microRNA-host-gene locus-encoded long noncoding RNAs. Wiley Interdiscip. Rev. RNA 2021, 12, e1625. [Google Scholar] [CrossRef] [PubMed]
- Morlando, M.; Ballarino, M.; Gromak, N.; Pagano, F.; Bozzoni, I.; Proudfoot, N.J. Primary microRNA transcripts are processed co-transcriptionally. Nat. Struct. Mol. Biol. 2008, 15, 902–909. [Google Scholar] [CrossRef]
- Kim, Y.K.; Kim, V.N. Processing of intronic microRNAs. EMBO J. 2007, 26, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Raveh, E.; Matouk, I.J.; Gilon, M.; Hochberg, A. The H19 Long non-coding RNA in cancer initiation, progression and metastasis—A proposed unifying theory. Mol. Cancer 2015, 14, 184. [Google Scholar] [CrossRef]
- Zheng, X.; Hu, J.; Yue, S.; Kristiani, L.; Kim, M.; Sauria, M.; Taylor, J.; Kim, Y.; Zheng, Y. Lamins Organize the Global Three-Dimensional Genome from the Nuclear Periphery. Mol. Cell 2018, 71, 802–815.e7. [Google Scholar] [CrossRef]
- Burke, B.; Stewart, C.L. The nuclear lamins: Flexibility in function. Nat. Rev. Mol. Cell Biol. 2013, 14, 13–24. [Google Scholar] [CrossRef]
- Delva, E.; Tucker, D.K.; Kowalczyk, A.P. The desmosome. Cold Spring Harb. Perspect. Biol. 2009, 1, a002543. [Google Scholar] [CrossRef]
- Kowalczyk, A.P.; Bornslaeger, E.A.; Borgwardt, J.E.; Palka, H.L.; Dhaliwal, A.S.; Corcoran, C.M.; Denning, M.F.; Green, K.J. The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin-plakoglobin complexes. J. Cell Biol. 1997, 139, 773–784. [Google Scholar] [CrossRef]
- Jin, M.; Fuller, G.G.; Han, T.; Yao, Y.; Alessi, A.F.; Freeberg, M.A.; Roach, N.P.; Moresco, J.J.; Karnovsky, A.; Baba, M.; et al. Glycolytic Enzymes Coalesce in G Bodies under Hypoxic Stress. Cell Rep. 2017, 20, 895–908. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.; Nelson, J.C.; Bend, E.G.; Rodriguez-Laureano, L.; Tueros, F.G.; Cartagenova, L.; Underwood, K.; Jorgensen, E.M.; Colon-Ramos, D.A. Glycolytic Enzymes Localize to Synapses under Energy Stress to Support Synaptic Function. Neuron 2016, 90, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Fuller, G.G.; Kim, J.K. Compartmentalization and metabolic regulation of glycolysis. J. Cell Sci. 2021, 134, jcs258469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ji, X.; Li, P.; Liu, C.; Lou, J.; Wang, Z.; Wen, W.; Xiao, Y.; Zhang, M.; Zhu, X. Liquid-liquid phase separation in biology: Mechanisms, physiological functions and human diseases. Sci. China Life Sci. 2020, 63, 953–985. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Tang, Y.; Wang, J.; Xiong, F.; Guo, C.; Wang, Y.; Zhang, S.; Gong, Z.; Wei, F.; Yang, L.; et al. Role of long non-coding RNAs in glucose metabolism in cancer. Mol. Cancer 2017, 16, 130. [Google Scholar] [CrossRef]
- Zhu, Y.; Jin, L.; Shi, R.; Li, J.; Wang, Y.; Zhang, L.; Liang, C.Z.; Narayana, V.K.; De Souza, D.P.; Thorne, R.F.; et al. The long noncoding RNA glycoLINC assembles a lower glycolytic metabolon to promote glycolysis. Mol. Cell 2022, 82, 542–554.e6. [Google Scholar] [CrossRef]
- Park, M.K.; Zhang, L.; Min, K.W.; Cho, J.H.; Yeh, C.C.; Moon, H.; Hormaechea-Agulla, D.; Mun, H.; Ko, S.; Lee, J.W.; et al. NEAT1 is essential for metabolic changes that promote breast cancer growth and metastasis. Cell Metab. 2021, 33, 2380–2397.e9. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Diepenbruck, M.; Tiede, S.; Saxena, M.; Ivanek, R.; Kalathur, R.K.R.; Luond, F.; Meyer-Schaller, N.; Christofori, G. miR-1199-5p and Zeb1 function in a double-negative feedback loop potentially coordinating EMT and tumour metastasis. Nat. Commun. 2017, 8, 1168. [Google Scholar] [CrossRef]
- Yang, L.; Duff, M.O.; Graveley, B.R.; Carmichael, G.G.; Chen, L.L. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011, 12, R16. [Google Scholar] [CrossRef] [Green Version]
- Vance, K.W.; Sansom, S.N.; Lee, S.; Chalei, V.; Kong, L.; Cooper, S.E.; Oliver, P.L.; Ponting, C.P. The long non-coding RNA Paupar regulates the expression of both local and distal genes. EMBO J. 2014, 33, 296–311. [Google Scholar] [CrossRef] [PubMed]
- Subtil-Rodriguez, A.; Reyes, J.C. BRG1 helps RNA polymerase II to overcome a nucleosomal barrier during elongation, in vivo. EMBO Rep. 2010, 11, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Quinn, J.; Chang, H.Y. Chromatin isolation by RNA purification (ChIRP). J. Vis. Exp. 2012, 61, e3912. [Google Scholar] [CrossRef]
- Jacobi, A.M.; Rettig, G.R.; Turk, R.; Collingwood, M.A.; Zeiner, S.A.; Quadros, R.M.; Harms, D.W.; Bonthuis, P.J.; Gregg, C.; Ohtsuka, M.; et al. Simplified CRISPR tools for efficient genome editing and streamlined protocols for their delivery into mammalian cells and mouse zygotes. Methods 2017, 121–122, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.P.; Cifuentes-Rojas, C.; Kesner, B.; Aeby, E.; Lee, H.G.; Wei, C.; Oh, H.J.; Boukhali, M.; Haas, W.; Lee, J.T. TERRA RNA Antagonizes ATRX and Protects Telomeres. Cell 2017, 170, 86–101.e16. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Gutierrez-Vazquez, C.; Sanchez-Cabo, F.; Perez-Hernandez, D.; Vazquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sanchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef]
- Lorenzo, C.; Delgado, P.; Busse, C.E.; Sanz-Bravo, A.; Martos-Folgado, I.; Bonzon-Kulichenko, E.; Ferrarini, A.; Gonzalez-Valdes, I.B.; Mur, S.M.; Roldan-Montero, R.; et al. ALDH4A1 is an atherosclerosis auto-antigen targeted by protective antibodies. Nature 2021, 589, 287–292. [Google Scholar] [CrossRef]
- Martinez-Bartolome, S.; Navarro, P.; Martin-Maroto, F.; Lopez-Ferrer, D.; Ramos-Fernandez, A.; Villar, M.; Garcia-Ruiz, J.P.; Vazquez, J. Properties of average score distributions of SEQUEST: The probability ratio method. Mol. Cell. Proteom. 2008, 7, 1135–1145. [Google Scholar] [CrossRef]
- Navarro, P.; Vazquez, J. A refined method to calculate false discovery rates for peptide identification using decoy databases. J. Proteome Res. 2009, 8, 1792–1796. [Google Scholar] [CrossRef]
- Bonzon-Kulichenko, E.; Garcia-Marques, F.; Trevisan-Herraz, M.; Vazquez, J. Revisiting peptide identification by high-accuracy mass spectrometry: Problems associated with the use of narrow mass precursor windows. J. Proteome Res. 2015, 14, 700–710. [Google Scholar] [CrossRef]
- Chernousov, M.A.; Stahl, R.C.; Carey, D.J. Schwann cell type V collagen inhibits axonal outgrowth and promotes Schwann cell migration via distinct adhesive activities of the collagen and noncollagen domains. J. Neurosci. 2001, 21, 6125–6135. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polo-Generelo, S.; Torres, B.; Guerrero-Martínez, J.A.; Camafeita, E.; Vázquez, J.; Reyes, J.C.; Pintor-Toro, J.A. TGF-β-Upregulated Lnc-Nr6a1 Acts as a Reservoir of miR-181 and Mediates Assembly of a Glycolytic Complex. Non-Coding RNA 2022, 8, 62. https://doi.org/10.3390/ncrna8050062
Polo-Generelo S, Torres B, Guerrero-Martínez JA, Camafeita E, Vázquez J, Reyes JC, Pintor-Toro JA. TGF-β-Upregulated Lnc-Nr6a1 Acts as a Reservoir of miR-181 and Mediates Assembly of a Glycolytic Complex. Non-Coding RNA. 2022; 8(5):62. https://doi.org/10.3390/ncrna8050062
Chicago/Turabian StylePolo-Generelo, Salvador, Belén Torres, José A. Guerrero-Martínez, Emilio Camafeita, Jesús Vázquez, José C. Reyes, and José A. Pintor-Toro. 2022. "TGF-β-Upregulated Lnc-Nr6a1 Acts as a Reservoir of miR-181 and Mediates Assembly of a Glycolytic Complex" Non-Coding RNA 8, no. 5: 62. https://doi.org/10.3390/ncrna8050062