

Genetic Deletion of the LINC00520 Homolog in Mouse Aggravates Angiotensin II-Induced Hypertension

 , , ,
, , ,  , and
, and 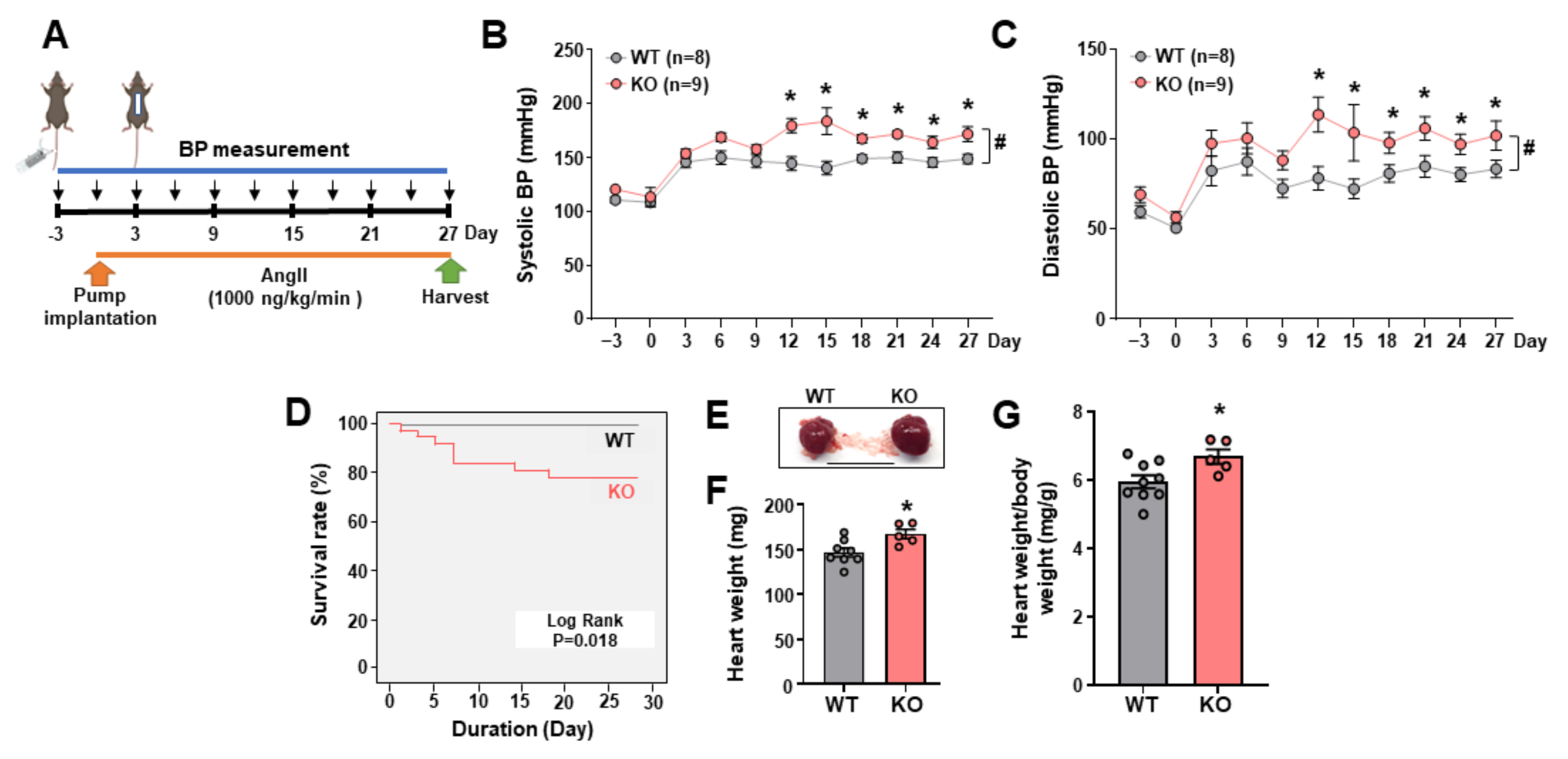{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Leene-Knockout (KO) Mice Develop Higher BP in an Angiotensin II (AngII)-Induced HTN Model
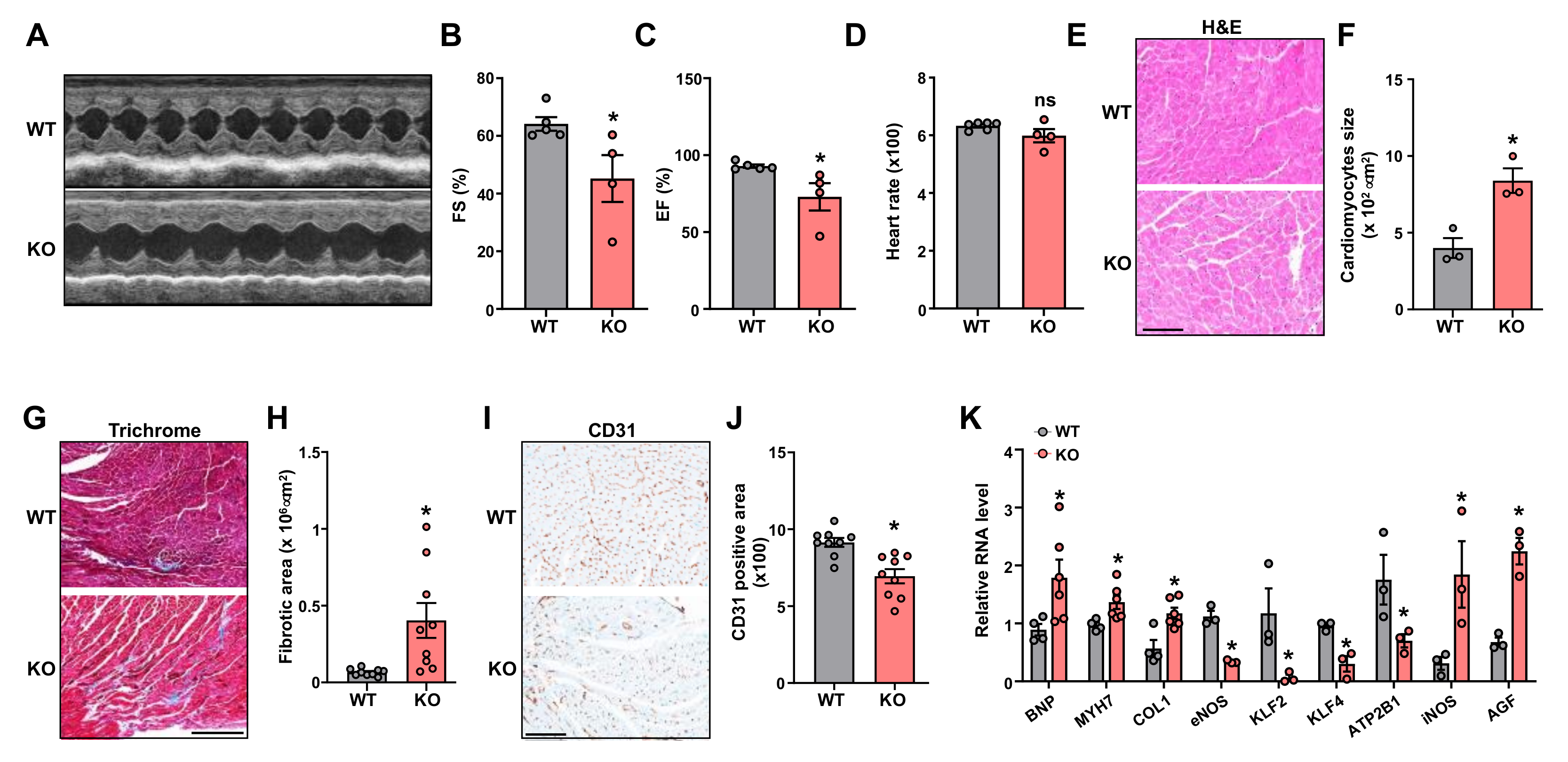
2.2. Leene-KO Aggravates AngII-Induced Cardiac Hypertrophy and Pathology
2.3. AngII Infused Leene-KO Mice Show Aggravated Kidney Damage
2.4. Potential Role of Endothelial LEENE in BP Regulation
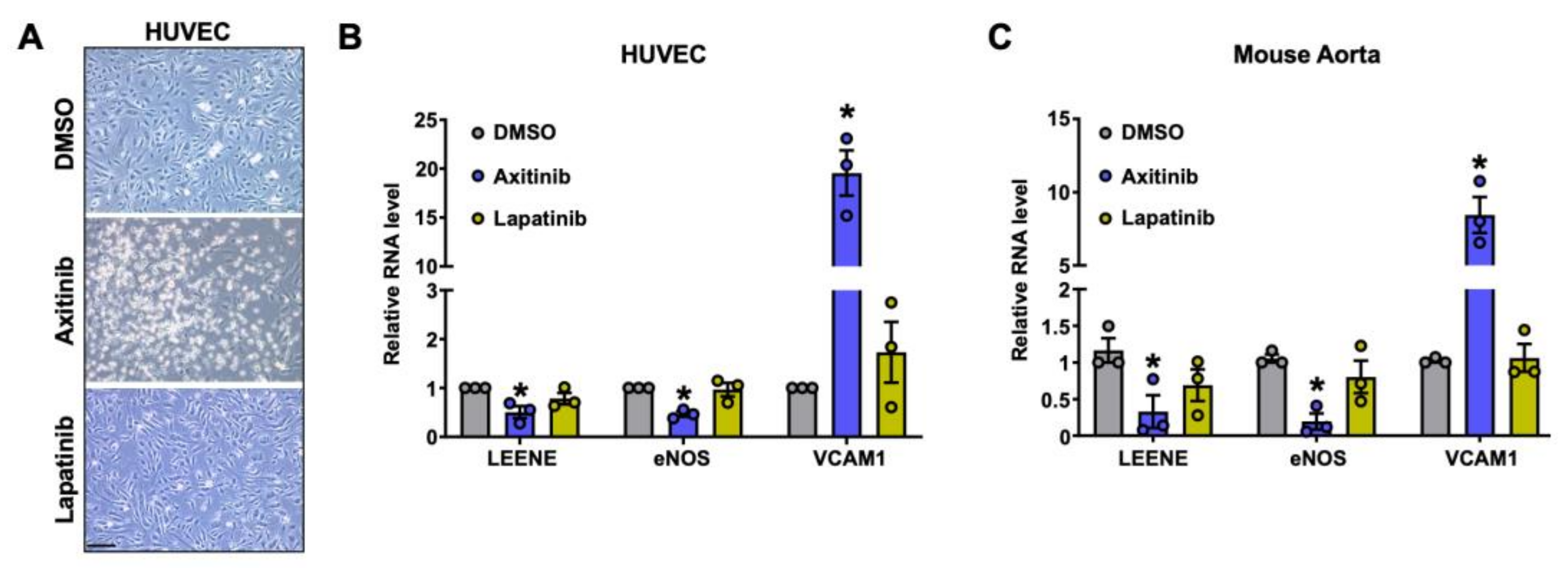
2.5. Effect of TKIs on LEENE Levels in ECs
3. Discussion
4. Materials and Methods
4.1. Mouse Model
4.2. Measurement of BP and Echocardiography
4.3. Histology and Immunostaining
4.4. Isolation of ECs from Murine Lungs and Aorta Ring Assay
4.5. RNA Extraction, Quantitative PCR (qPCR), and RNA-Seq Library Preparation and Analysis
4.6. HUVEC Culture and Drug Treatment
4.7. RNA IP (RIP)
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Worldwide trends in blood pressure from 1975 to 2015: A pooled analysis of 1479 population-based measurement studies with 19·1 million participants. Lancet 2017, 389, 37–55. [CrossRef] [PubMed] [Green Version]
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Carretero, O.A.; Oparil, S. Essential hypertension. Part I: Definition and etiology. Circulation 2000, 101, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Bavishi, C.; Bangalore, S.; Messerli, F.H. Outcomes of Intensive Blood Pressure Lowering in Older Hypertensive Patients. J. Am. Coll. Cardiol. 2017, 69, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.S.; Masi, S.; Taddei, S. Understanding the role of genetics in hypertension. Eur. Heart J. 2017, 38, 2309–2312. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.D.; Newton-Cheh, C.; Chasman, D.I.; Ehret, G.B.; Johnson, T.; Rose, L.; Rice, K.; Verwoert, G.C.; Launer, L.J.; Gudnason, V.; et al. Association of hypertension drug target genes with blood pressure and hypertension in 86,588 individuals. Hypertension 2011, 57, 903–910. [Google Scholar] [CrossRef]
- Padmanabhan, S.; Caulfield, M.; Dominiczak, A.F. Genetic and molecular aspects of hypertension. Circ. Res. 2015, 116, 937–959. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, T.J.; Ehret, G.B.; Nandakumar, P.; Ranatunga, D.; Schaefer, C.; Kwok, P.Y.; Iribarren, C.; Chakravarti, A.; Risch, N. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat. Genet. 2017, 49, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, C.P.; Ng, F.L.; Warren, H.R.; Barnes, M.R.; Munroe, P.B.; Caulfield, M.J. Exploring hypertension genome-wide association studies findings and impact on pathophysiology, pathways, and pharmacogenetics. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 73–90. [Google Scholar] [CrossRef]
- Wilson, C.; Zhang, X.; Buckley, C.; Heathcote, H.R.; Lee, M.D.; McCarron, J.G. Increased Vascular Contractility in Hypertension Results From Impaired Endothelial Calcium Signaling. Hypertension 2019, 74, 1200–1214. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, F. Long noncoding RNA: A new contributor and potential therapeutic target in fibrosis. Epigenomics 2017, 9, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Shah, R.; Dimmeler, S.; Freedman, J.E.; Holley, C.; Lee, J.M.; Moore, K.; Musunuru, K.; Wang, D.Z.; Xiao, J.; et al. Noncoding RNAs in Cardiovascular Disease: Current Knowledge, Tools and Technologies for Investigation, and Future Directions: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2020, 13, e000062. [Google Scholar] [CrossRef] [PubMed]
- Sallam, T.; Sandhu, J.; Tontonoz, P. Long Noncoding RNA Discovery in Cardiovascular Disease: Decoding Form to Function. Circ. Res. 2018, 122, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Bretschneider, T.; Kittner, K.H. Combined modality treatment of malignant glioma in adults. Radiobiol. Radiother. 1986, 27, 287–295. [Google Scholar]
- Samara, V.A.; Das, S.; Reddy, M.A.; Tanwar, V.S.; Stapleton, K.; Leung, A.; Abdollahi, M.; Ganguly, R.; Lanting, L.; Natarajan, R. Angiotensin II-Induced Long Non-Coding RNA Alivec Regulates Chondrogenesis in Vascular Smooth Muscle Cells. Cells 2021, 10, 2696. [Google Scholar] [CrossRef]
- Das, S.; Zhang, E.; Senapati, P.; Amaram, V.; Reddy, M.A.; Stapleton, K.; Leung, A.; Lanting, L.; Wang, M.; Chen, Z.; et al. A Novel Angiotensin II-Induced Long Noncoding RNA Giver Regulates Oxidative Stress, Inflammation, and Proliferation in Vascular Smooth Muscle Cells. Circ. Res. 2018, 123, 1298–1312. [Google Scholar] [CrossRef]
- Das, S.; Senapati, P.; Chen, Z.; Reddy, M.A.; Ganguly, R.; Lanting, L.; Mandi, V.; Bansal, A.; Leung, A.; Zhang, S.; et al. Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells. Nat. Commun. 2017, 8, 1467. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Jose, P.A.; Zeng, C. Noncoding RNAs in the Regulatory Network of Hypertension. Hypertension 2018, 72, 1047–1059. [Google Scholar] [CrossRef]
- Jin, L.; Lin, X.; Yang, L.; Fan, X.; Wang, W.; Li, S.; Li, J.; Liu, X.; Bao, M.; Cui, X.; et al. AK098656, a Novel Vascular Smooth Muscle Cell-Dominant Long Noncoding RNA, Promotes Hypertension. Hypertension 2018, 71, 262–272. [Google Scholar] [CrossRef]
- Song, J.; Huang, S.; Wang, K.; Li, W.; Pao, L.; Chen, F.; Zhao, X. Long Non-coding RNA MEG3 Attenuates the Angiotensin II-Induced Injury of Human Umbilical Vein Endothelial Cells by Interacting With p53. Front. Genet. 2019, 10, 78. [Google Scholar] [CrossRef]
- Bu, S.; Nguyen, H.C.; Michels, D.C.R.; Rasheed, B.; Nikfarjam, S.; Singh, R.; Wang, L.; Patel, D.A.; Singh, S.; Qadura, M.; et al. Transcriptomics of angiotensin II-induced long noncoding and coding RNAs in endothelial cells. J. Hypertens. 2022, 40, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Ajami, N.E.; Huang, T.S.; Lin, F.M.; Lou, C.H.; Wang, Y.T.; Li, S.; Kang, J.; Munkacsi, H.; Maurya, M.R.; et al. Enhancer-associated long non-coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat. Commun. 2018, 9, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Luo, Y.; Yuan, D.; Calandrelli, R.; Malhi, N.K.; Sriram, K.; Miao, Y.; Lou, C.H.; Tsark, W.; Tapia, A.; et al. Long noncoding RNA LEENE promotes angiogenesis and ischemic recovery in diabetes models. J. Clin. Investig. 2023, 133, e161759. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J.; Antman, E.M.; Black, H.R.; Hayes, D.L.; Manson, J.E.; Plutzky, J.; Popma, J.J.; Stevenson, W. The cardiovascular disease continuum validated: Clinical evidence of improved patient outcomes: Part I: Pathophysiology and clinical trial evidence (risk factors through stable coronary artery disease). Circulation 2006, 114, 2850–2870. [Google Scholar] [CrossRef] [PubMed]
- Crowley, S.D.; Gurley, S.B.; Herrera, M.J.; Ruiz, P.; Griffiths, R.; Kumar, A.P.; Kim, H.S.; Smithies, O.; Le, T.H.; Coffman, T.M. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc. Natl. Acad. Sci. USA 2006, 103, 17985–17990. [Google Scholar] [CrossRef] [Green Version]
- Pacanowski, M.A.; Zineh, I.; Cooper-Dehoff, R.M.; Pepine, C.J.; Johnson, J.A. Genetic and pharmacogenetic associations between NOS3 polymorphisms, blood pressure, and cardiovascular events in hypertension. Am. J. Hypertens. 2009, 22, 748–753. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.; Yu, M.Y.; Moon, J.J.; Kim, J.E.; Lee, S.; Han, S.W.; Park, D.J.; Kim, Y.S.; Yang, S.H. Renoprotective Effect of KLF2 on Glomerular Endothelial Dysfunction in Hypertensive Nephropathy. Cells 2022, 11, 762. [Google Scholar] [CrossRef]
- Shatat, M.A.; Tian, H.; Zhang, R.; Tandon, G.; Hale, A.; Fritz, J.S.; Zhou, G.; Martínez-González, J.; Rodríguez, C.; Champion, H.C.; et al. Endothelial Krüppel-like factor 4 modulates pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2014, 50, 647–653. [Google Scholar] [CrossRef] [Green Version]
- Tabara, Y.; Kohara, K.; Kita, Y.; Hirawa, N.; Katsuya, T.; Ohkubo, T.; Hiura, Y.; Tajima, A.; Morisaki, T.; Miyata, T.; et al. Common variants in the ATP2B1 gene are associated with susceptibility to hypertension: The Japanese Millennium Genome Project. Hypertension 2010, 56, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Mattson, D.L.; Maeda, C.Y.; Bachman, T.D.; Cowley, A.W., Jr. Inducible nitric oxide synthase and blood pressure. Hypertension 1998, 31, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Townsend, R.R. Chemotherapy agents and hypertension: A focus on angiogenesis blockade. Curr. Hypertens. Rep. 2007, 9, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Herrmann, J. Cardiotoxicity with vascular endothelial growth factor inhibitor therapy. NPJ Precis. Oncol. 2018, 2, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Luo, S.; Peng, G.; Lu, J.Y.; Cui, G.; Liu, L.; Yan, P.; Yin, Y.; Liu, W.; Wang, R.; et al. Mouse knockout models reveal largely dispensable but context-dependent functions of lncRNAs during development. J. Mol. Cell Biol. 2018, 10, 175–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humar, R.; Zimmerli, L.; Battegay, E. Angiogenesis and hypertension: An update. J. Hum. Hypertens. 2009, 23, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Mongiardi, M.P.; Radice, G.; Piras, M.; Stagni, V.; Pacioni, S.; Re, A.; Putti, S.; Ferrè, F.; Farsetti, A.; Pallini, R.; et al. Axitinib exposure triggers endothelial cells senescence through ROS accumulation and ATM activation. Oncogene 2019, 38, 5413–5424. [Google Scholar] [CrossRef]
- Xue, B.; Pamidimukkala, J.; Hay, M. Sex differences in the development of angiotensin II-induced hypertension in conscious mice. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2177–H2184. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Singh, M.; Guo, F.; Hay, M.; Johnson, A.K. Protective actions of estrogen on angiotensin II-induced hypertension: Role of central nitric oxide. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1638–H1646. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Miao, Y.; Luo, Y.; Sriram, K.; Qi, Z.; Lin, F.M.; Gu, Y.; Lai, C.H.; Hsu, C.Y.; Peterson, K.L.; et al. Suppression of Endothelial AGO1 Promotes Adipose Tissue Browning and Improves Metabolic Dysfunction. Circulation 2020, 142, 365–379. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, X.; Li, C.; Li, Q.; An, Y.A.; Luo, X.; Deng, Y.; Gillette, T.G.; Scherer, P.E.; Wang, Z.V. Integrated Stress Response Couples Mitochondrial Protein Translation with Oxidative Stress Control. Circulation 2021, 144, 1500–1515. [Google Scholar] [CrossRef]
- Bellacen, K.; Lewis, E.C. Aortic ring assay. J. Vis. Exp. JoVE 2009, 24, 1564. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, X.; Lai, C.-H.; Malhi, N.K.; Chadha, R.; Luo, Y.; Liu, X.; Yuan, D.; Tapia, A.; Abdollahi, M.; Zhang, G.; et al. Genetic Deletion of the LINC00520 Homolog in Mouse Aggravates Angiotensin II-Induced Hypertension. Non-Coding RNA 2023, 9, 31. https://doi.org/10.3390/ncrna9030031
Tang X, Lai C-H, Malhi NK, Chadha R, Luo Y, Liu X, Yuan D, Tapia A, Abdollahi M, Zhang G, et al. Genetic Deletion of the LINC00520 Homolog in Mouse Aggravates Angiotensin II-Induced Hypertension. Non-Coding RNA. 2023; 9(3):31. https://doi.org/10.3390/ncrna9030031
Chicago/Turabian StyleTang, Xiaofang, Chih-Hung Lai, Naseeb K. Malhi, Rahuljeet Chadha, Yingjun Luo, Xuejing Liu, Dongqiang Yuan, Alonso Tapia, Maryam Abdollahi, Guangyu Zhang, and et al. 2023. "Genetic Deletion of the LINC00520 Homolog in Mouse Aggravates Angiotensin II-Induced Hypertension" Non-Coding RNA 9, no. 3: 31. https://doi.org/10.3390/ncrna9030031
APA StyleTang, X., Lai, C.-H., Malhi, N. K., Chadha, R., Luo, Y., Liu, X., Yuan, D., Tapia, A., Abdollahi, M., Zhang, G., Calandrelli, R., Shiu, Y.-T., Wang, Z. V., Rhee, J.-W., Zhong, S., Natarajan, R., & Chen, Z. B. (2023). Genetic Deletion of the LINC00520 Homolog in Mouse Aggravates Angiotensin II-Induced Hypertension. Non-Coding RNA, 9(3), 31. https://doi.org/10.3390/ncrna9030031