
Theoretical Studies on the Dynamical Behavior of Atom/Ion Migration on the Surface of Pristine and BN-Doped Graphene
Abstract
:
1. Introduction
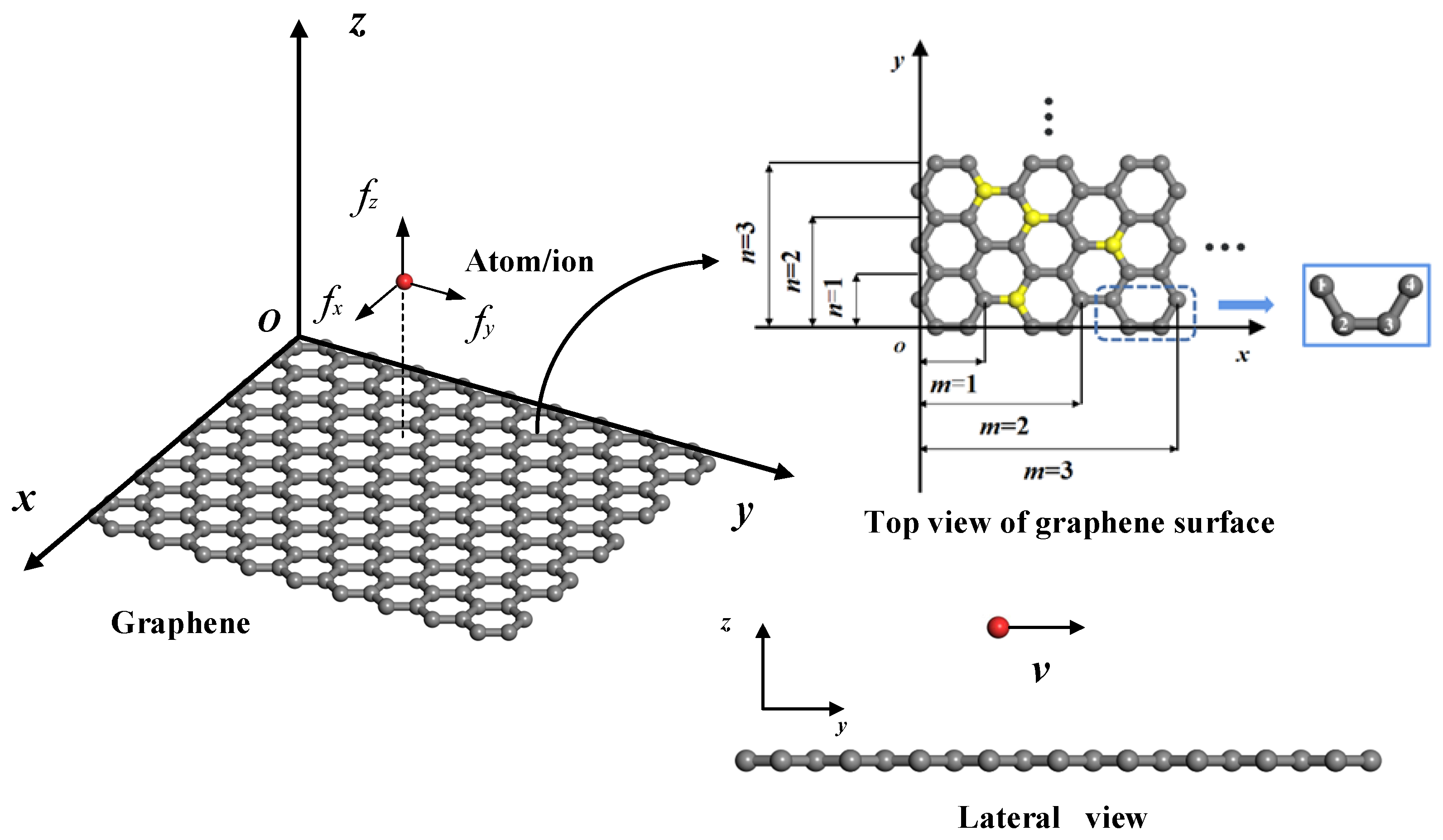
2. Theoretical Model and Method
3. Results and Discussion
3.1. Migration of Atoms/Ions along Specific Paths Inside a Finite-Size Graphene Surface
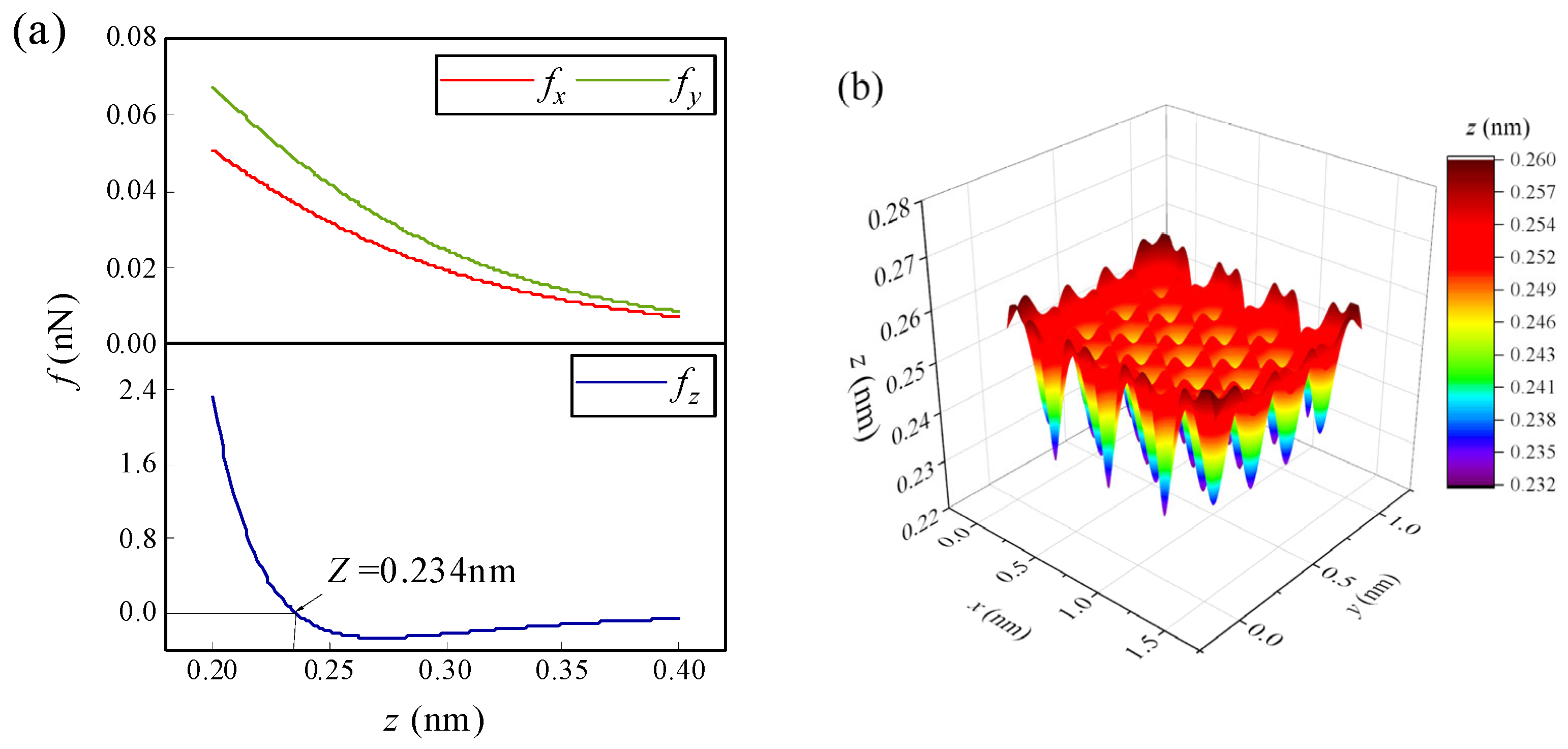
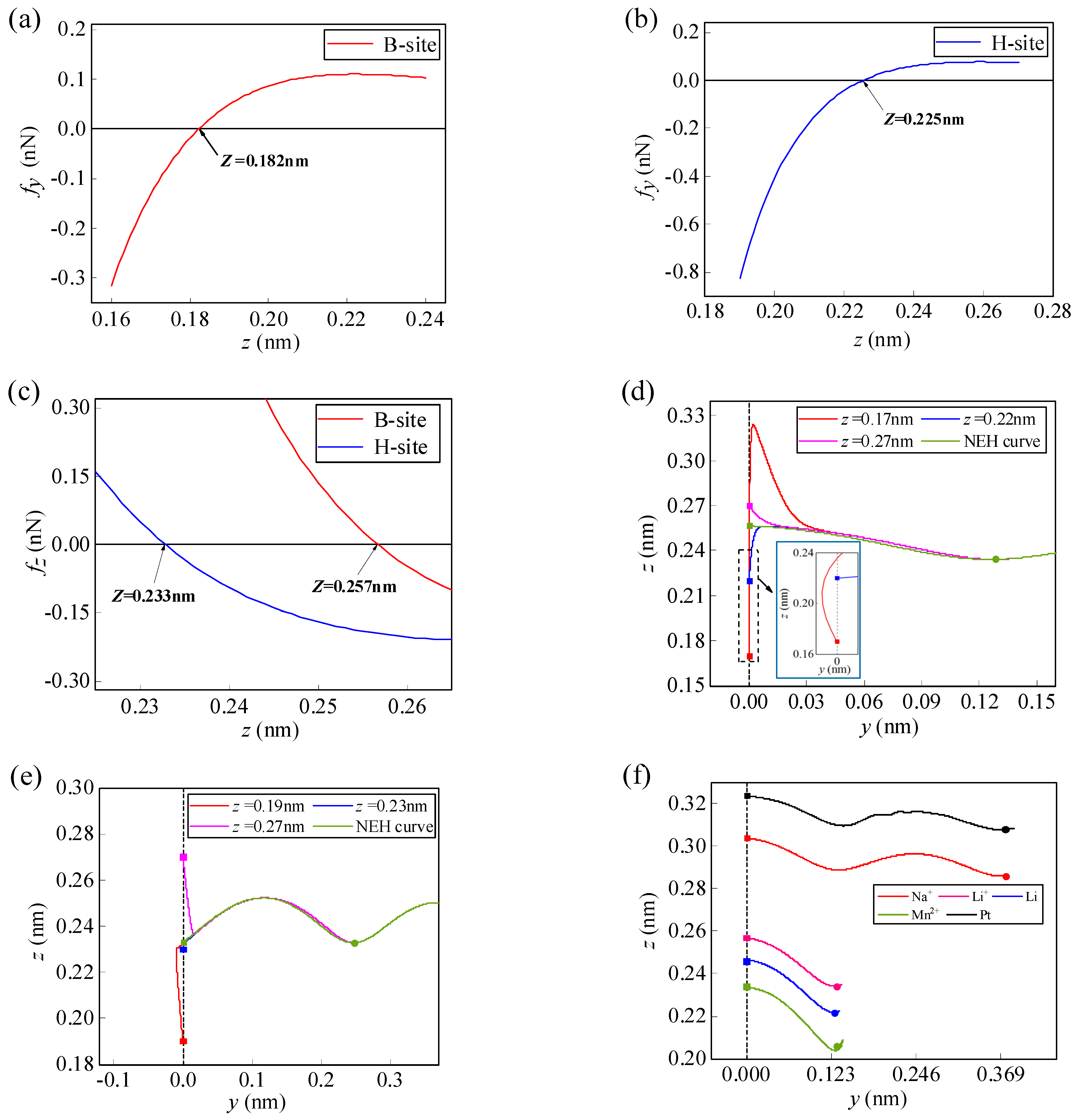
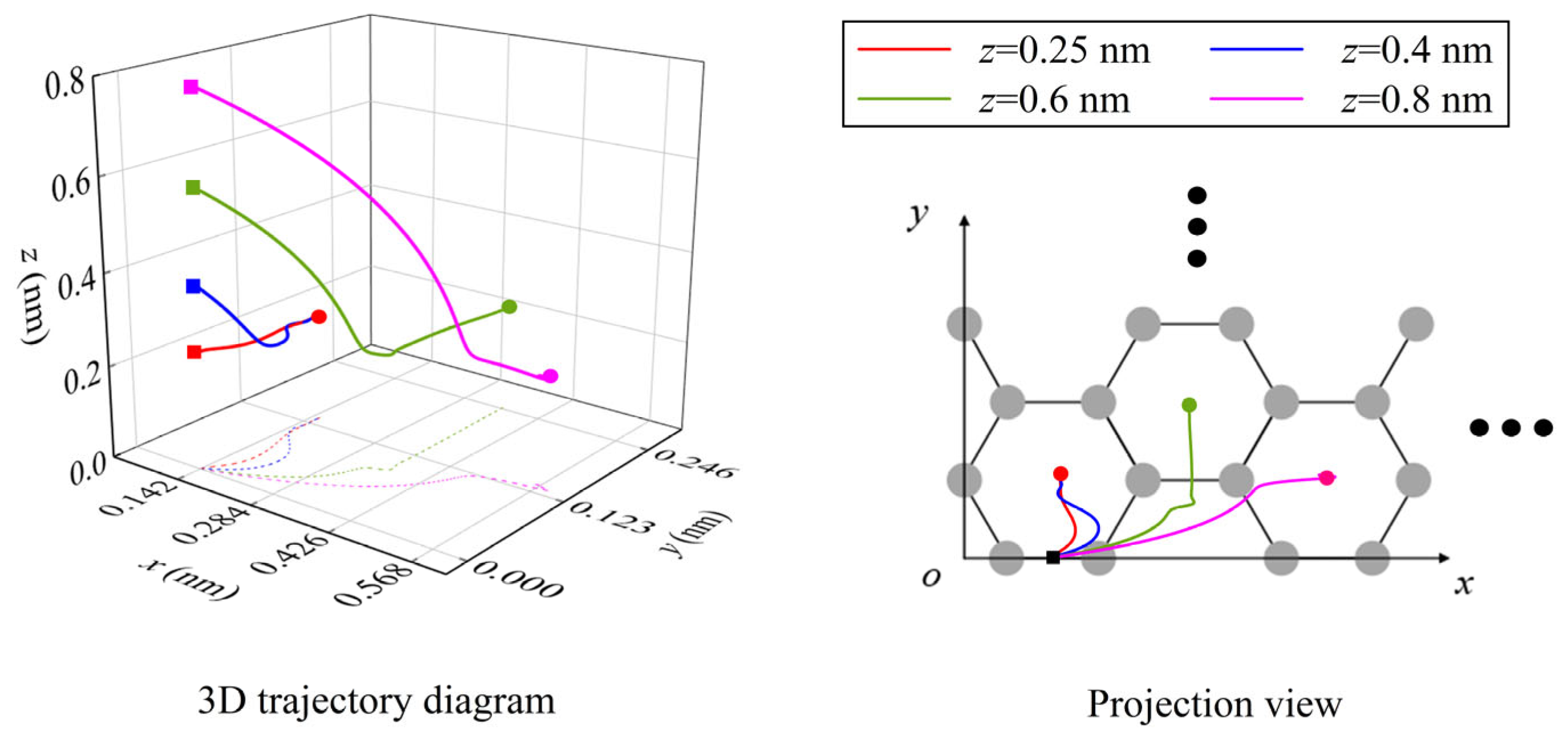
3.1.1. Effect of Initial Height on the Spontaneous Migration of Atoms/Ions
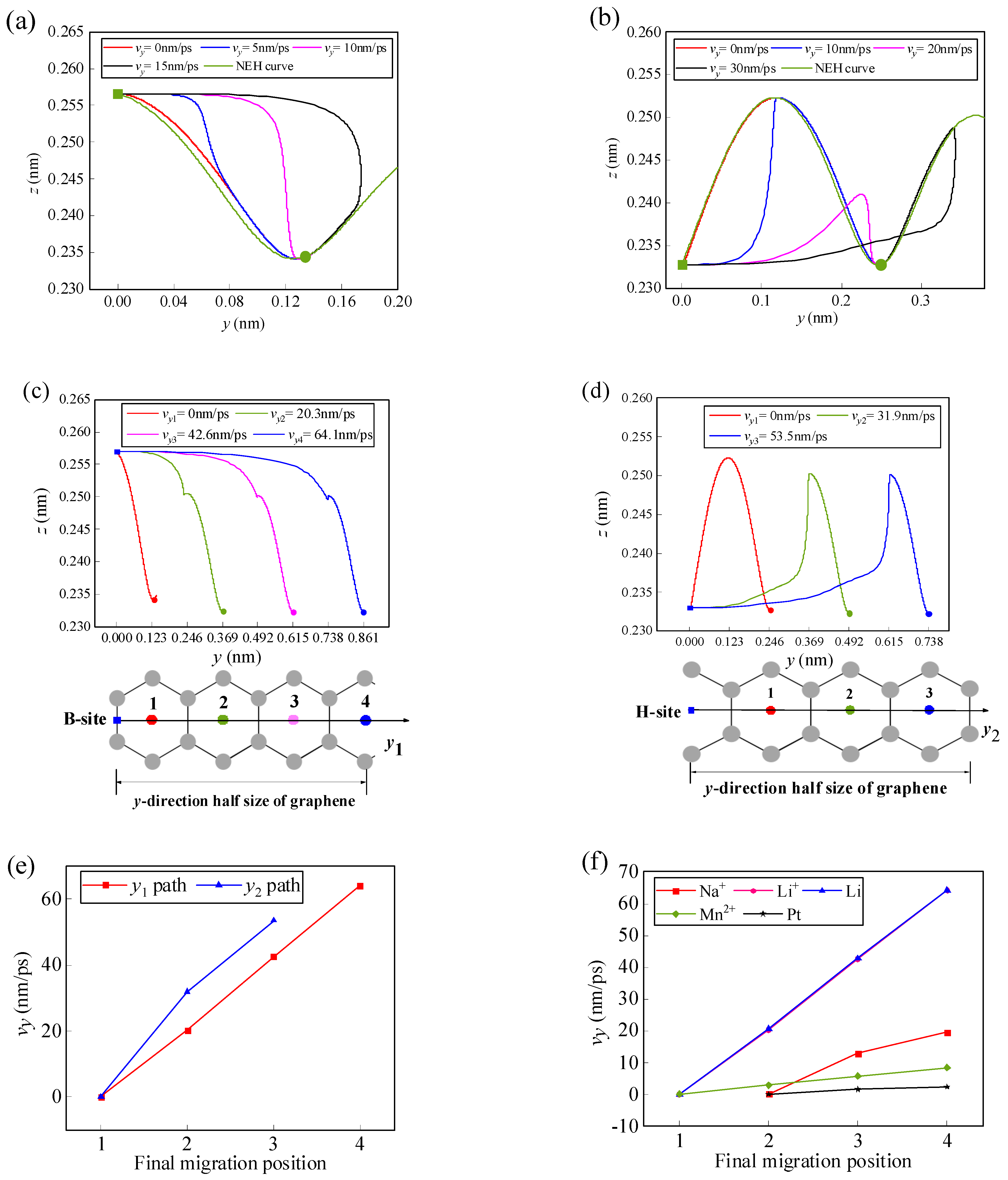
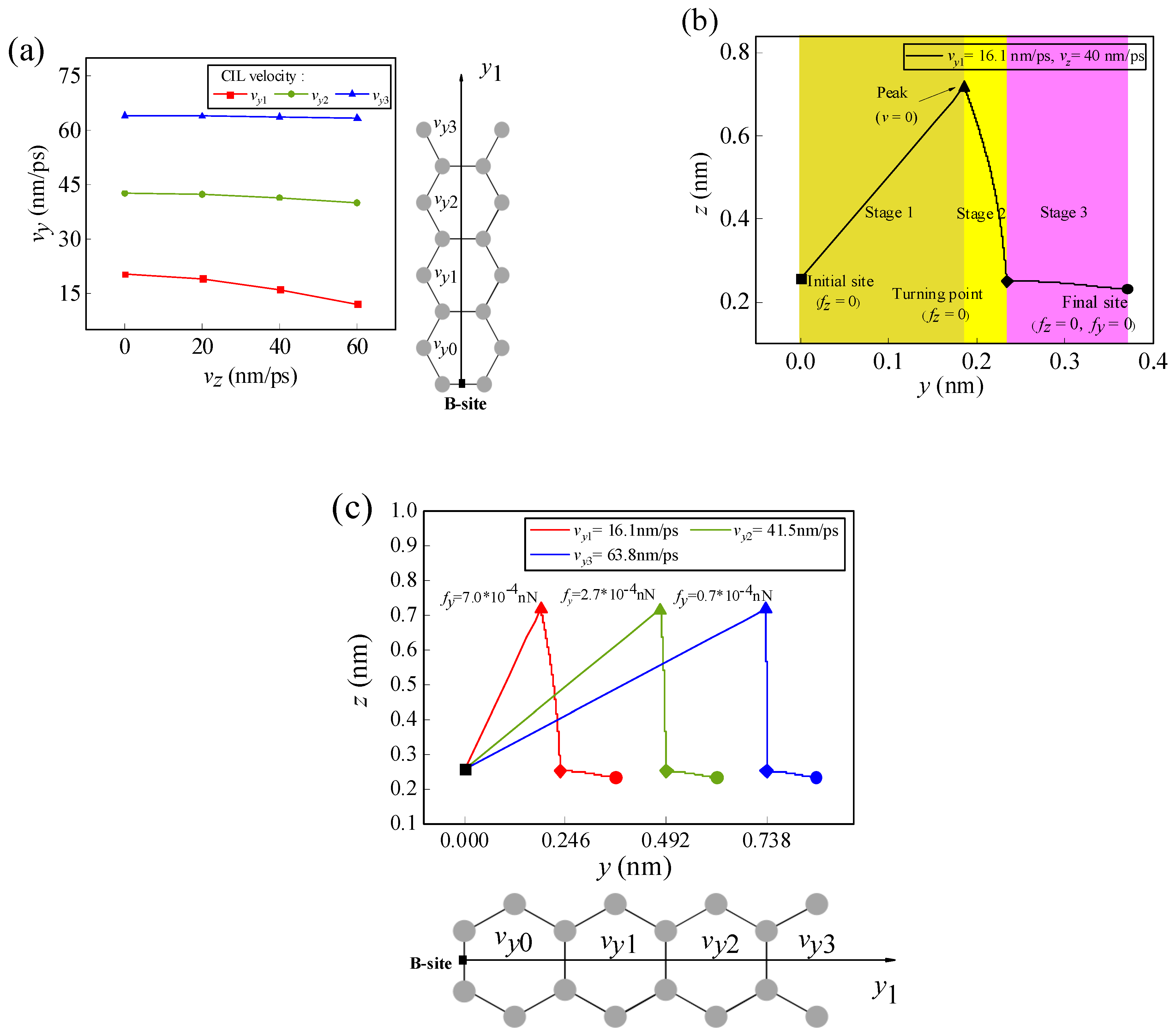
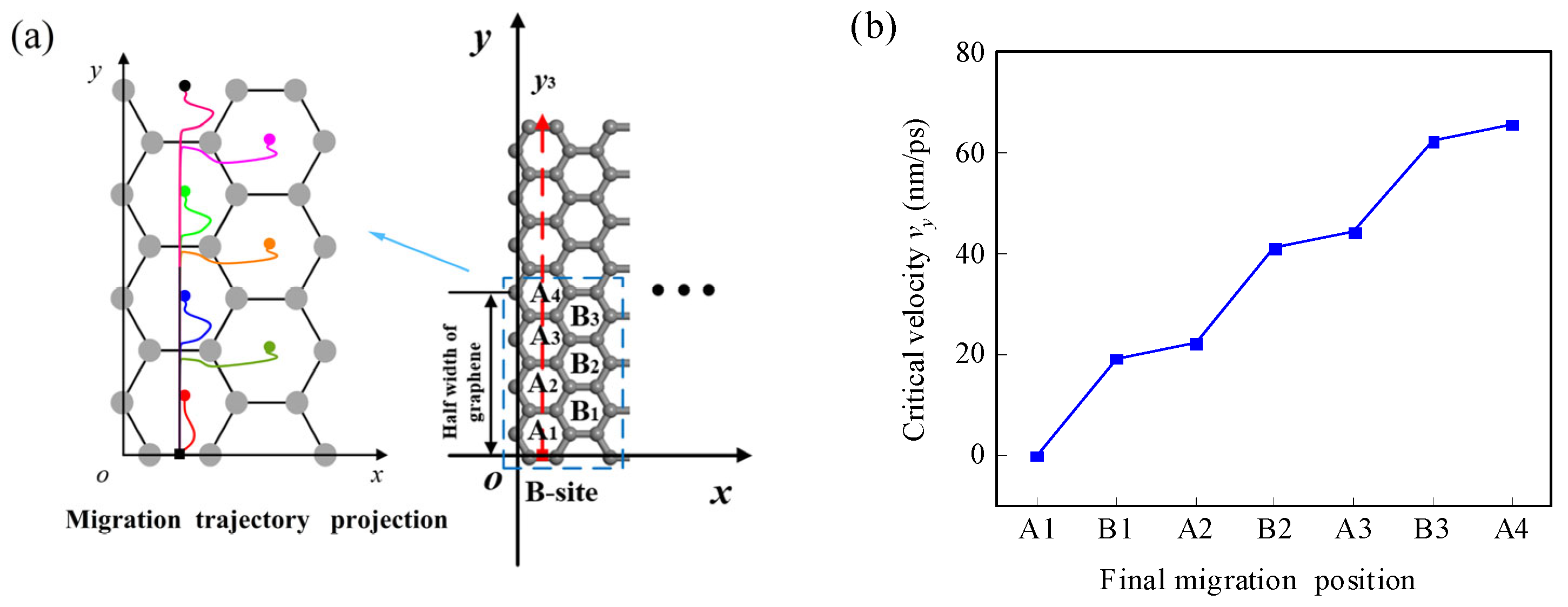
3.1.2. Effect of Initial Velocity on Atom/Ion Migration
3.2. Migration of the Li-Ion along the Specific Path at the Edge of the Graphene Surface
3.2.1. Effect of Initial Height on the Spontaneous Migration of Li-Ions
3.2.2. Effect of Initial Velocity on the Migration of the Li-Ion
3.3. Regulation of BN Doping on Li-Ion Migration Behavior
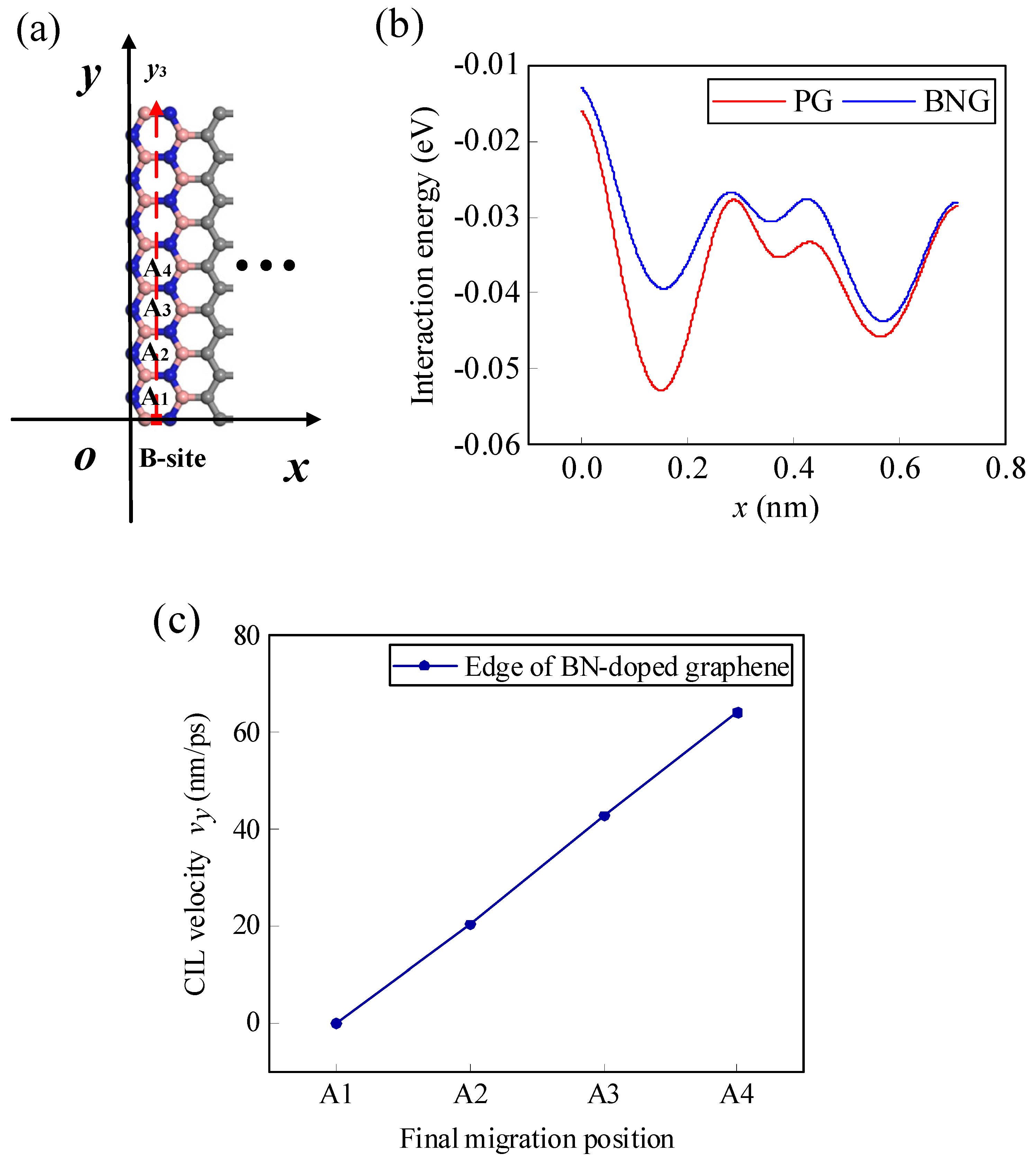
3.3.1. Effect of BN Doping on the Migration Behavior of the Li-Ion along a Specific Internal Path
3.3.2. Regulation of BN Doping on Li-ion Migration Behavior along a Specific Path at the Edge
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, Y.L.; Xie, B.; Zhang, Z.; Zheng, Q.S.; Xu, Z.P. Mechanical properties of graphene papers. J. Mech. Phys. Solids 2012, 60, 591–605. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, K.; Dixit, A.R. A review of the mechanical and thermal properties of graphene and its hybrid polymer nanocomposites for structural applications. J. Mater. Sci. 2019, 54, 5992–6026. [Google Scholar] [CrossRef]
- Young, R.J.; Kinloch, I.A.; Gong, L.; Novoselov, K.S. The mechanics of graphene nanocomposites: A review. Compos. Sci. Technol. 2012, 72, 1459–1476. [Google Scholar] [CrossRef]
- Wang, J.; Chen, B.L. Adsorption and coadsorption of organic pollutants and a heavy metal by graphene oxide and reduced graphene materials. Chem. Eng. J. 2015, 281, 379–388. [Google Scholar] [CrossRef]
- Liu, X.; Sun, J.; Xu, X.T.; Alsaedi, A.; Hayat, T.; Li, J.X. Adsorption and desorption of U (VI) on different-size graphene oxide. Chem. Eng. J. 2019, 360, 941–950. [Google Scholar] [CrossRef]
- Bu, J.Q.; Yuan, L.; Zhang, N.; Liu, D.; Meng, Y.; Peng, X. High-efficiency adsorption of methylene blue dye from wastewater by a thiosemicarbazide functionalized graphene oxide composite. Diam. Relat. Mater. 2020, 101, 107604. [Google Scholar] [CrossRef]
- Suarez-Iglesias, O.; Collado, S.; Oulego, P.; Diaz, M. Graphene-family nanomaterials in wastewater treatment plants. Chem. Eng. J. 2017, 313, 121–135. [Google Scholar] [CrossRef]
- Chatterjee, S.G.; Chatterjee, S.; Ray, A.K.; Chakraborty, A.K. Graphene-metal oxide nanohybrids for toxic gas sensor: A review. Sens. Actuator B Chem. 2015, 221, 1170–1181. [Google Scholar] [CrossRef]
- Ye, M.H.; Zhang, Z.P.; Zhao, Y.; Qu, L.T. Graphene platforms for smart energy generation and storage. Joule 2018, 2, 245–268. [Google Scholar] [CrossRef]
- Wong, D.; Corsetti, F.; Wang, Y.; Brar, V.W.; Tsai, H.Z.; Wu, Q.; Kawakami, R.K.; Zettl, A.; Mostofi, A.A.; Lischner, J.; et al. Spatially resolving density-dependent screening around a single charged atom in graphene. Phys. Rev. B 2017, 95, 205419. [Google Scholar] [CrossRef]
- Ersan, G.; Apul, O.G.; Perreault, F.; Karanfil, T. Adsorption of organic contaminants by graphene nanosheets: A review. Water Res. 2017, 126, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Velusamy, S.; Roy, A.; Sundaram, S.; Mallick, T.K. A review on heavy metal ions and containing dyes removal through graphene oxide-based adsorption strategies for textile wastewater treatment. Chem. Rec. 2021, 21, 1570–1610. [Google Scholar] [CrossRef] [PubMed]
- Hosseingholipourasl, A.; Ariffin, S.H.S.; Al-Otaibi, Y.D.; Akbari, E.; Hamid, F.K.H.; Koloor, S.S.R.; Petrů, M. Analytical approach to study sensing properties of graphene based gas sensor. Sensors 2020, 20, 1506. [Google Scholar] [CrossRef]
- Yu, D.Y.W.; Prikhodchenko, P.V.; Mason, C.W.; Batabyal, S.K.; Gun, J.; Sladkevich, S.; Medvedev, A.G.; Lev, O. High-capacity antimony sulphide nanoparticle-decorated graphene composite as anode for sodium-ion batteries. Nat. Commun. 2013, 4, 2922. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, J.G. Theoretical characterization and application of mechanical behavior of atoms/ions migrating on graphene surface. Mater. Chem. Phys. 2021, 260, 124138. [Google Scholar] [CrossRef]
- Gan, Y.J.; Sun, L.T.; Banhart, F. One- and two-dimensional diffusion of metal atoms in graphene. Small 2008, 4, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xiong, W.; Zhou, Y.S.; Lu, Y.F.; Zeng, X.C. An ab initio study of the nickel- catalyzed transformation of amorphous carbon into graphene in rapid thermal processing. Nanoscale 2016, 8, 9746–9755. [Google Scholar] [CrossRef]
- Yao, F.; Günes, F.; Ta, H.Q.; Lee, S.M.; Chae, S.J.; Sheem, K.Y.; Cojocaru, C.S.; Xie, S.S.; Lee, Y.H. Diffusion mechanism of lithium ion through basal plane of layered graphene. J. Am. Chem. Soc. 2012, 134, 8646–8654. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.Z.; Guo, J.G.; Zhou, L.J. Theoretical investigation on the adsorption and diffusion of lithium-ion on and between graphene layers with size and defect effects. Adsorpt. Sci. Technol. 2016, 34, 212. [Google Scholar] [CrossRef]
- Li, M.; Guo, J.G. Theoretical study for adsorption and migration behavior of atoms/ions on sinusoidal corrugated graphene surface. Mater. Chem. Phys. 2020, 254, 123527. [Google Scholar] [CrossRef]
- Leggesse, E.G.; Chen, C.L.; Jiang, J.C. Lithium diffusion in graphene and graphite: Effect of edge morphology. Carbon 2016, 103, 209–216. [Google Scholar] [CrossRef]
- Xian, L.; Chou, M.Y. Diffusion of Si and C atoms on and between graphene layers. J. Phys. D Appl. Phys. 2012, 45, 455309. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, L.T.; Xu, Z.J.; Krasheninnikov, A.V.; Huai, P.; Zhu, Z.Y.; Banhart, F. Migration of gold atoms in graphene ribbons: Role of the edges. Phys. Rev. B 2010, 81, 125425. [Google Scholar] [CrossRef]
- Tang, Y.N.; Yang, Z.X.; Dai, X.Q. Trapping of metal atoms in the defects on graphene. J. Chem. Phys. 2011, 135, 224704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.H.; Zhao, J. The effect of carbon defects on Cl anion diffusion in graphene based on first principles calculations. Mater. Res. Express 2017, 4, 075604. [Google Scholar] [CrossRef]
- Tachikawa, H.; Kawabata, H. DFT and direct MD study of the diffusion of sodium ion on graphenes. Thin Solid Films 2009, 518, 873–876. [Google Scholar] [CrossRef]
- Tachikawa, H.; Iyama, T.; Kawabata, H. Effects of fluorine atom substitution of graphene edge site on the diffusion of lithium ion. Jpn. J. Appl. Phys. 2010, 49, 01AH02. [Google Scholar] [CrossRef]
- Han, D.; Wang, X.; Zhao, Y.B.; Chen, Y.; Tang, M.X.; Zhao, Z.Q. High-quality graphene synthesis on amorphous SiC through a rapid thermal treatment. Carbon 2017, 124, 105–110. [Google Scholar] [CrossRef]
- Mishra, N.; Boeckl, J.; Motta, N.; Iacopi, F. Graphene growth on silicon carbide: A review. Phys. Status Solidi A 2016, 213, 2277–2289. [Google Scholar] [CrossRef]
- Chen, K.; Sun, Z.H.; Fang, R.P.; Shi, Y.; Cheng, H.M.; Li, F. Metal-organic frameworks (MOFs)-derived nitrogen-doped porous carbon anchored on graphene with multifunctional effects for lithium-sulfur batteries. Adv. Funct. Mater. 2018, 28, 1707592. [Google Scholar] [CrossRef]
- Nong, W.; Liang, H.K.; Qin, S.H.; Li, Y.; Wang, C.X. Computational design of two-dimensional boron-containing compounds as efficient metal-free electrocatalysts toward nitrogen reduction independent of heteroatom doping. ACS Appl. Mater. Inter. 2020, 12, 50505–505015. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.L.; Na, N.; Mohamadi, A. Investigation the application of pristine graphdiyne (GDY) and boron-doped graphdiyne (BGDY) as an electronic sensor for detection of anticancer drug. Comput. Theor. Chem. 2020, 1190, 112996. [Google Scholar] [CrossRef]
- Li, J.; Lin, L.; Rui, D.; Li, Q.; Zhang, J.; Kang, N.; Zhang, Y.; Peng, H.; Liu, Z.; Xu, H.Q. Electron–hole symmetry breaking in charge transport in nitrogen-doped graphene. ACS Nano 2017, 11, 4641–4650. [Google Scholar] [CrossRef] [PubMed]
- Marconcini, P.; Cresti, A.; Roche, S. Effect of the channel length on the transport characteristics of transistors based on boron-doped graphene ribbons. Materials 2018, 11, 667. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Shen, Z.; Liu, A.Q.; Kuo, J.-L. Band gap opening of graphene by doping small boron nitride domains. Nanoscale 2012, 4, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.K.; Guo, J.G.; Zhou, L.J. Theoretical studies on adsorption and migration of atoms/ions on the surface of boron, nitrogen and boron nitride doped graphene. Appl. Phys. A 2022, 128, 887. [Google Scholar] [CrossRef]
- Nachimuthu, S.; Lai, P.J.; Leggesse, E.G.; Jiang, J.C. A first principles study on boron-doped graphene decorated by Ni-Ti-Mg atoms for enhanced hydrogen storage performance. Sci. Rep. 2015, 5, 16797. [Google Scholar] [CrossRef]
- Das, D.; Kim, S.; Lee, K.R.; Singh, A.K. Li diffusion through doped and defected graphene. Phys. Chem. Chem. Phys. 2013, 15, 15128–15134. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.H.; Ren, Z.Y.; Wan, L.J.; Zheng, J.M.; Guo, P.; Zhou, Y.X. Density functional theory prediction for diffusion of lithium on boron-doped graphene surface. Appl. Surf. Sci. 2011, 257, 7443–7446. [Google Scholar] [CrossRef]
- Lee, H.W.; Moon, H.S.; Hur, J.; Kim, I.T.; Park, M.S.; Yun, J.M.; Kim, K.H.; Lee, S.G. Mechanism of sodium adsorption on N-doped graphene nanoribbons for sodium ion battery applications: A density functional theory approach. Carbon 2017, 119, 492–501. [Google Scholar] [CrossRef]
- Petrushenko, I.K.; Petrushenko, K.B. Adsorption of diatomic molecules on graphene, h-BN and their BNC heterostructures: DFT study. Diam. Relat. Mater. 2019, 100, 107575. [Google Scholar] [CrossRef]
- Ornstein, R.L.; Rein, R.; Breen, D.L.; Macelroy, R.D. An optimized potential function for the calculation of nucleic acid interaction energies I. Base stacking. Biopolymers 1978, 17, 2341–2360. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, H.; Schwarz, U.D.; Wiesendanger, R. Modelling of the scan process in lateral force microscopy. Surf. Sci. 1997, 375, 395–402. [Google Scholar] [CrossRef]
- Zheng, J.M.; Ren, Z.Y.; Guo, P.; Fang, L.; Fan, J. Diffusion of Li+ ion on graphene: A DFT study. Appl. Surf. Sci. 2011, 258, 1651–1655. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NEH (nm) | Na+ | Li+ | Li | Mn2+ | Pt |
|---|---|---|---|---|---|
| Initial B-site | 0.304 | 0.257 | 0.246 | 0.234 | 0.324 |
| Initial H-site | 0.287 | 0.233 | 0.221 | 0.205 | 0.309 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.-K.; Zhou, L.-J.; Guo, J.-G. Theoretical Studies on the Dynamical Behavior of Atom/Ion Migration on the Surface of Pristine and BN-Doped Graphene. C 2024, 10, 59. https://doi.org/10.3390/c10030059
Zhang T-K, Zhou L-J, Guo J-G. Theoretical Studies on the Dynamical Behavior of Atom/Ion Migration on the Surface of Pristine and BN-Doped Graphene. C. 2024; 10(3):59. https://doi.org/10.3390/c10030059
Chicago/Turabian StyleZhang, Tong-Kun, Li-Jun Zhou, and Jian-Gang Guo. 2024. "Theoretical Studies on the Dynamical Behavior of Atom/Ion Migration on the Surface of Pristine and BN-Doped Graphene" C 10, no. 3: 59. https://doi.org/10.3390/c10030059