
The X-ray, Raman and TEM Signatures of Cellulose-Derived Carbons Explained

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Experimental
2.1. Materials, Treatments, and Terminology
2.2. X-ray Diffraction
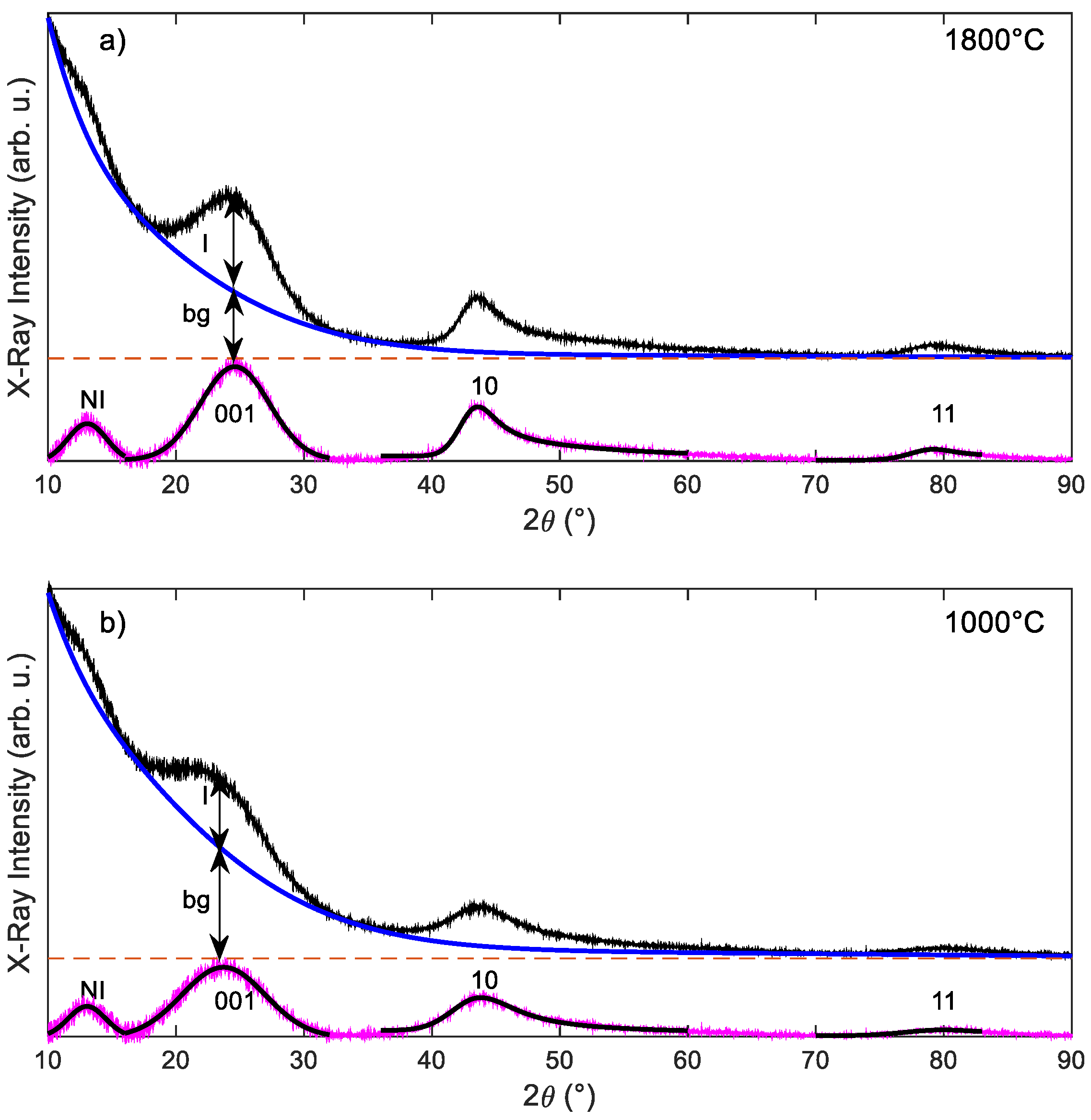
2.3. Modelling the X-ray Diffraction Peak at 2θ ≈ 13°
2.4. Raman Spectroscopy
2.4.1. Fitting Procedure and Parameters When Considering the D and G Bands Only
2.4.2. Fitting Procedure and Parameters When Considering Five Bands
2.5. HRTEM Micrographs, Image Filtering Analysis, Fringe Analysis
3. Results and Discussion
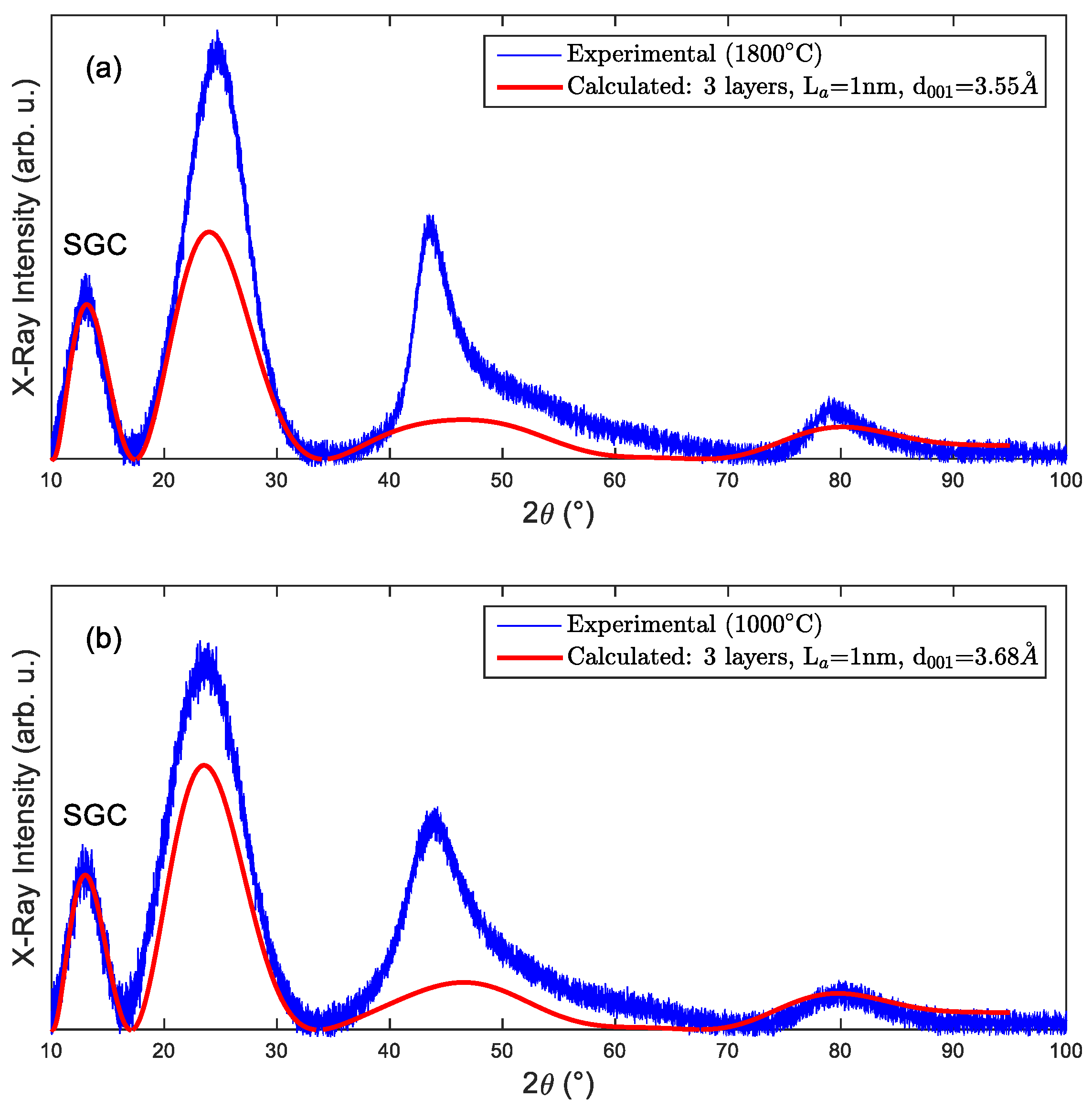
3.1. X-ray Diffraction
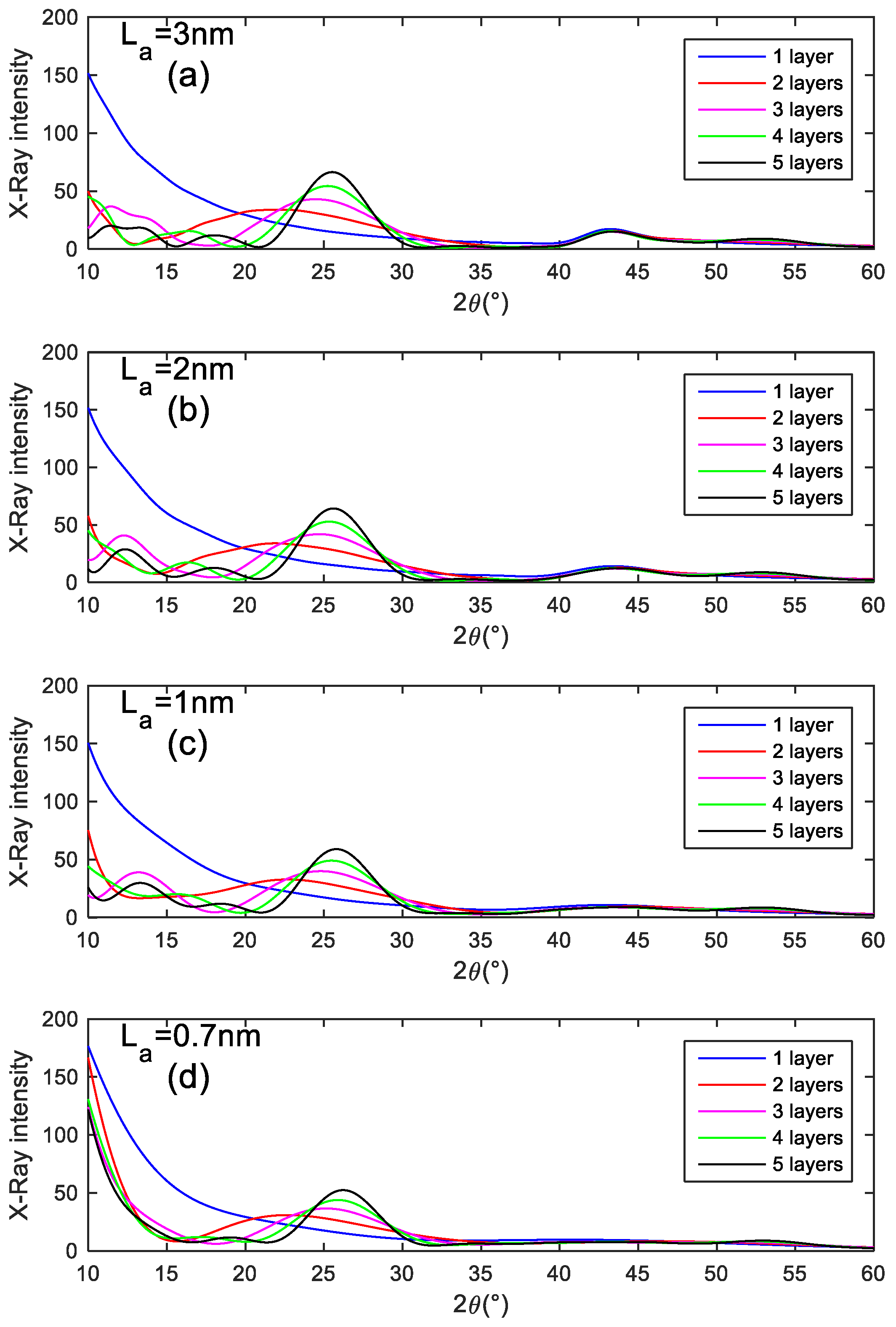
- Single graphenes generate a large intensity at a low 2θ angle whatever La, rapidly decreasing as the scattering angle increases. This is consistent with a recent statement from the literature, which showed that poorly organized carbon is able to contribute significantly to the background, specifically at low angles [40]. Therefore, our modelling supports this statement, and identifies that such a contributing intensity profile at low angles reveals the occurrence of individual graphenes.
- For an odd number (here 3 and 5) of turbostratically stacked layers with La in the range 1–2 nm, a peak shows up in the 13° range, resulting from interference effects. The position of this peak is dependent on La. Stacks involving an even number of graphenes (here 2 and 4) do not exhibit this peak, meaning that, in this case, interferences are destructive. This destructive effect rapidly vanishes as the number of graphenes in the stacks increases beyond 5.
- The actual 001 peak resulting from the stacking periodicity shift towards lower angles (from ~25.5 to ~22°) while the number of graphenes in the stack decreases (here from 5 to 2), as shown by Fujimoto [10], despite the fact that the intergraphene distance remains the same in our simulation.
- With ultra-small La (<1 nm), intensity is generated at a low 2θ angle which can significantly contribute to the background.
3.2. Raman Spectroscopy
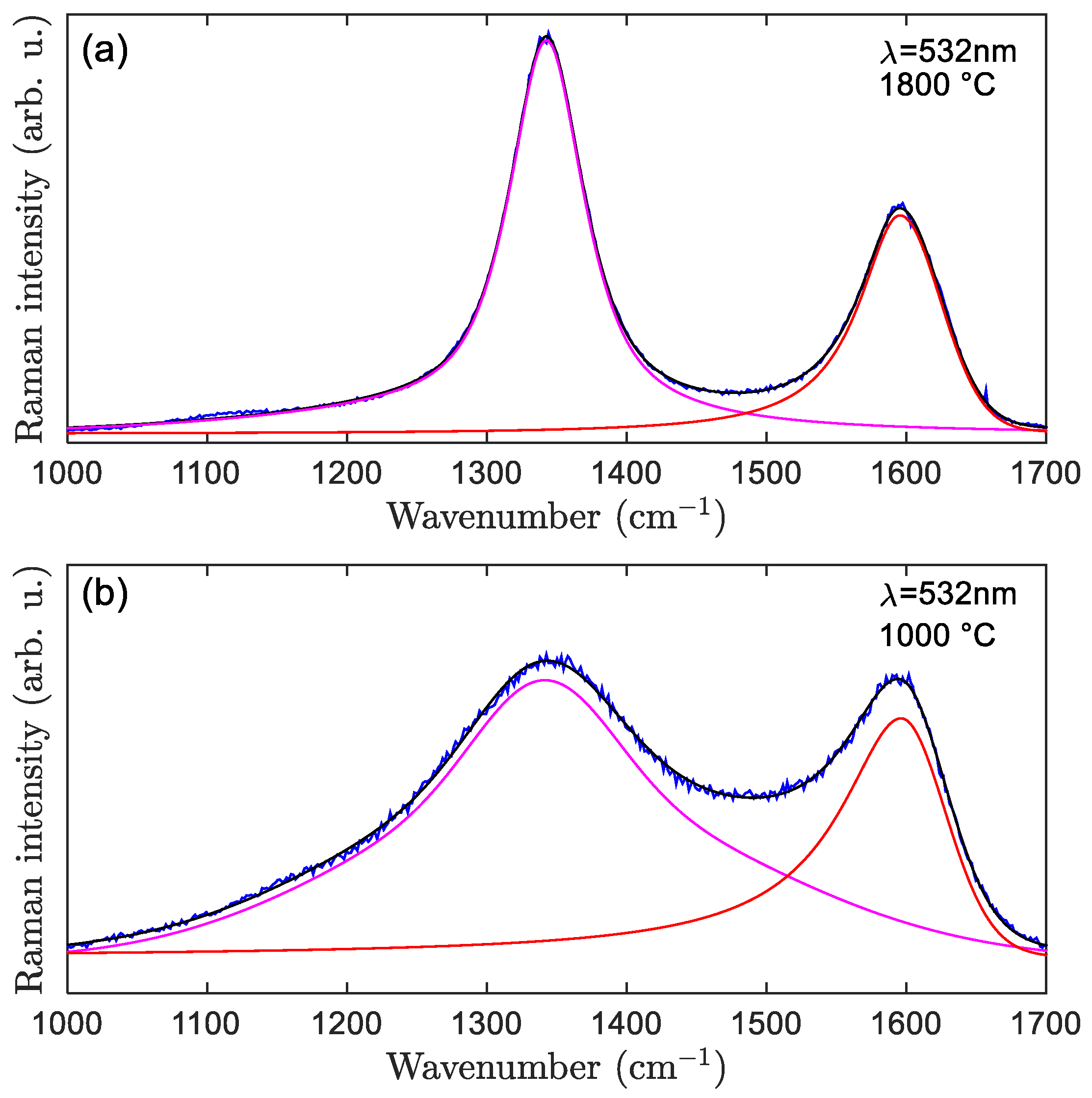
3.2.1. Raman Spectra Fitted with Two Bands (D and G Only)
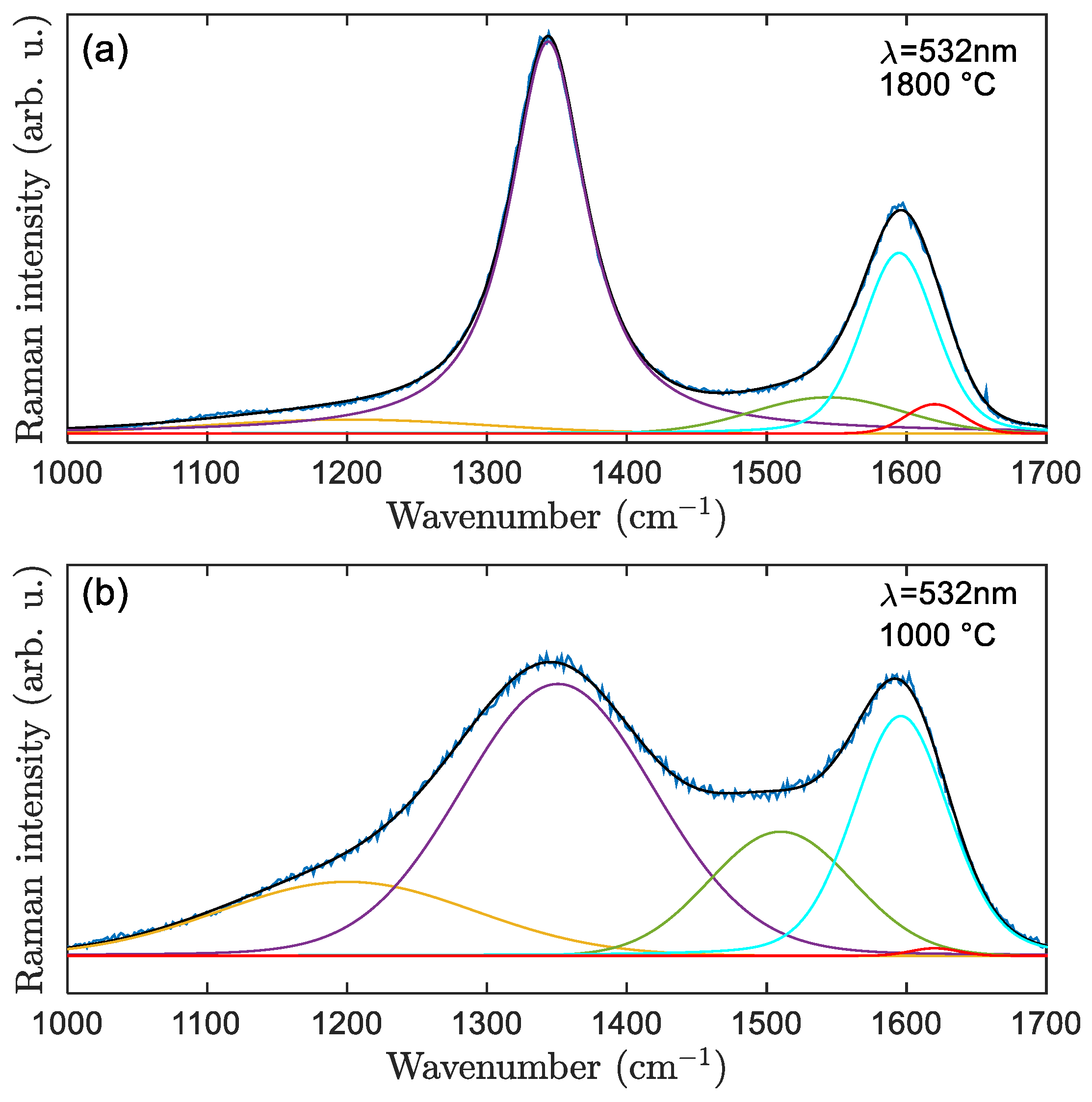
3.2.2. Raman Spectra Fitted with 5 Bands
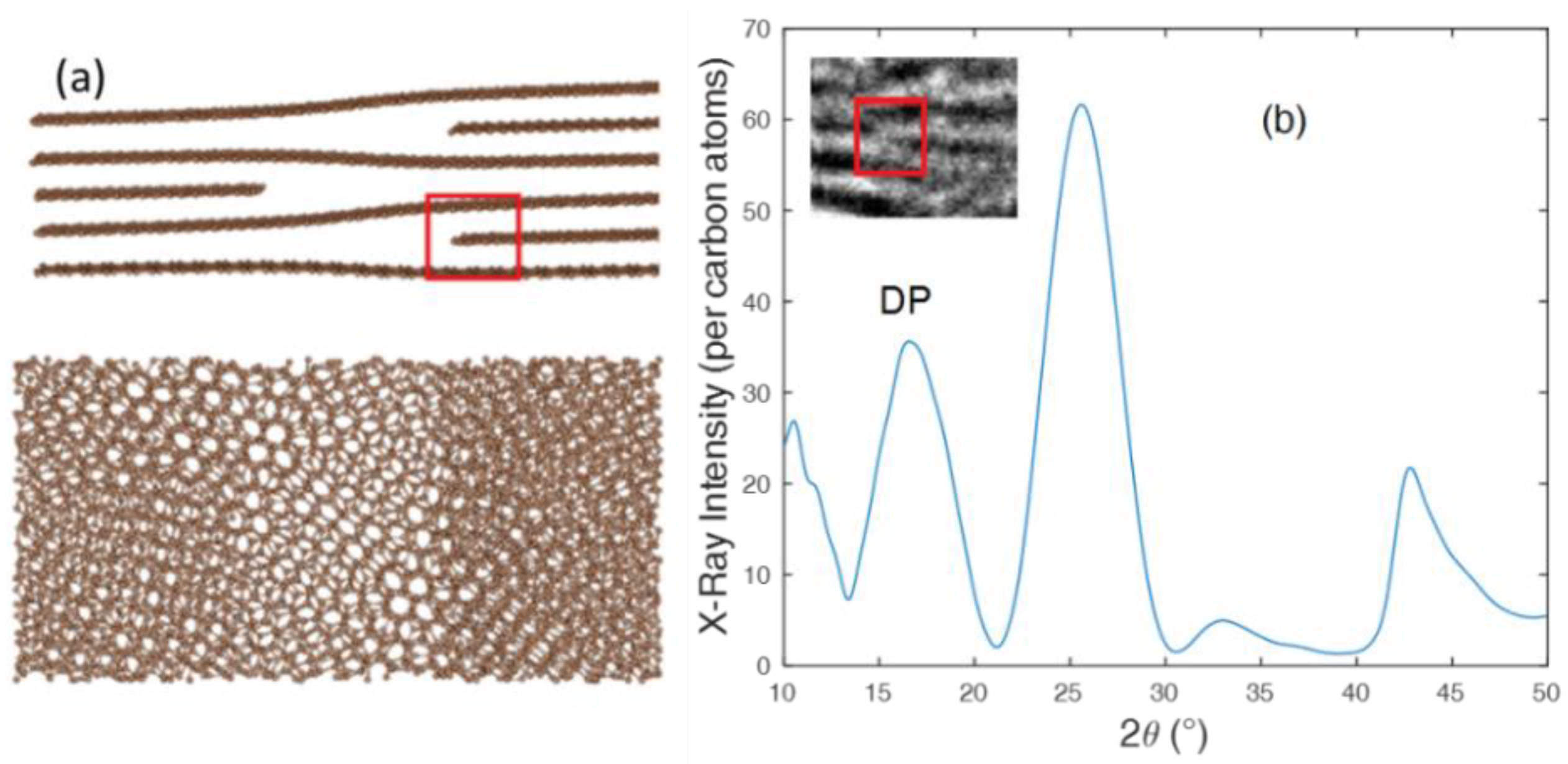
3.3. HRTEM Image Filtering Analysis, Fringe Analysis
4. Merging All the Data
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chowdhury, Z.; Abd Hamid, S.B.; Das, R.; Hasan, M.R.; Zain, S.; Khalisanni, K.; Uddin, M. Preparation of Carbonaceous Adsorbents from Lignocellulosic Biomass and Their Use in Removal of Contaminants from Aqueous Solution. BioResources 2013, 8, 6523–6555. [Google Scholar] [CrossRef]
- Mohamad Nor, N.; Lau, L.C.; Lee, K.T.; Mohamed, A.R. Synthesis of activated carbon from lignocellulosic biomass and its applications in air pollution control—A review. J. Environ. Chem. Eng. 2013, 1, 658–666. [Google Scholar] [CrossRef]
- Gan, L.; Zhu, J.; Lv, L. Cellulose hydrolysis catalyzed by highly acidic lignin-derived carbonaceous catalyst synthesized via hydrothermal carbonization. Cellulose 2017, 24, 5327–5339. [Google Scholar] [CrossRef]
- Zhang, P.; Gong, Y.; Li, H.; Chen, Z.; Wang, Y. Solvent-free aerobic oxidation of hydrocarbons and alcohols with Pd@N-doped carbon from glucose. Nat. Commun. 2013, 4, 1593. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, S.; Li, H.; Zan, Y.; Li, X.; Zhu, Y.; Dou, M.; Wang, F. Sustainable Carbonaceous Materials Derived from Biomass as Metal-Free Electrocatalysts. Adv. Mater. 2019, 31, e1805718. [Google Scholar] [CrossRef]
- Herou, S.; Schlee, P.; Jorge, A.B.; Titirici, M. Biomass-derived electrodes for flexible supercapacitors. Curr. Opin. Green Sustain. Chem. 2018, 9, 18–24. [Google Scholar] [CrossRef]
- Li, R.; Zhou, Y.; Li, W.; Zhu, J.; Huang, W. Structure Engineering in Biomass-Derived Carbon Materials for Electrochemical Energy Storage. Research 2020, 2020, 8685436. [Google Scholar] [CrossRef]
- Oberlin, A. Carbonization and graphitization. Carbon 1984, 22, 521–541. [Google Scholar] [CrossRef]
- Monthioux, M. Comments on: “Structure evolution mechanism of highly ordered graphite during carbonization of cellulose nanocrystals” by Eom et al. [Carbon 150 (2019) 142–152]. Carbon 2020, 160, 405–406. [Google Scholar] [CrossRef]
- Fujimoto, H. Theoretical X-ray scattering intensity of carbons with turbostratic stacking and AB stacking structures. Carbon 2003, 41, 1585–1592. [Google Scholar] [CrossRef]
- Puech, P.; Dabrowska, A.; Ratel-Ramond, N.; Vignoles, G.L.; Monthioux, M. New insight on carbonisation and graphitisation mechanisms as obtained from a bottom-up analytical approach of X-ray diffraction patterns. Carbon 2019, 147, 602–611. [Google Scholar] [CrossRef]
- Franklin, R.E.; Randall, J.T. Crystallite growth in graphitizing and non-graphitizing carbons. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1951, 209, 196–218. [Google Scholar]
- Gutierrez-Pardo, A.; Ramirez-Rico, J.; Cabezas-Rodríguez, R.; Fernández, J. Effect of catalytic graphitization on the electrochemical behavior of wood derived carbons for use in supercapacitors. J. Power Sources 2015, 278, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Maire, J.; Mering, J. Graphitization of soft carbons. Chem. Phys. Carbon 1970, 6, 125–190. [Google Scholar]
- Sembiring, S.; Situmeang, R.; Sembiring, Z. Synthesis and characterization of asphalt composite precursors using amorphous rice husk silica. Cerâmica 2019, 65, 194–199. [Google Scholar] [CrossRef]
- Cançado, L.G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y.A.; Mizusaki, H.; Jorio, A.; Coelho, L.N.; Magalhães-Paniago, R.; Pimenta, M.A. General equation for the determination of the crystallite size La of nanographite by Raman spectroscopy. Appl. Phys. Lett. 2006, 88, 163106. [Google Scholar] [CrossRef]
- Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S.; Cançado, L.G.; Jorio, A.; Saito, R. Studying disorder in graphite-based systems by Raman spectroscopy. Phys. Chem. Chem. Phys. 2007, 9, 1276–1290. [Google Scholar] [CrossRef]
- Puech, P.; Kandara, M.; Paredes, G.; Moulin, L.; Weiss-Hortala, E.; Kundu, A.; Ratel-Ramond, N.; Plewa, J.-M.; Pellenq, R.; Monthioux, M. Analyzing the Raman Spectra of Graphenic Carbon Materials from Kerogens to Nanotubes: What Type of Information Can Be Extracted from Defect Bands? C 2019, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Tuinstra, F.; Koenig, J. Raman Spectrum of Graphite. J. Chem. Phys. 1970, 53, 1126–1130. [Google Scholar] [CrossRef] [Green Version]
- Sadezky, A.; Muckenhuber, H.; Grothe, H.; Niessner, R.; Pöschl, U. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 8, 1731–1742. [Google Scholar] [CrossRef]
- Zickler, G.; Smarsly, B.; Gierlinger, N.; Peterlik, H.; Paris, O. A Reconsideration of the Relationship Between the Crystallite Size La of Carbons Determined by X-ray Diffraction and Raman Spectroscopy. Carbon 2006, 44, 3239–3246. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar] [CrossRef] [Green Version]
- Ager, J.W.; Veirs, D.K.; Shamir, J.; Rosenblatt, G.M. Laser heating effects in the characterization of carbon fibers by Raman spectroscopy. J. Appl. Phys. 1990, 68, 3598–3608. [Google Scholar] [CrossRef] [Green Version]
- Matthews, M.J.; Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S.; Endo, M. Origin of dispersive effects of the Raman D band in carbon materials. Phys. Rev. B 1999, 59, R6585–R6588. [Google Scholar] [CrossRef]
- Cançado, L.G.; Da Silva, M.G.; Ferreira, E.H.M.; Hof, F.; Kampioti, K.; Huang, K.; Jorio, A. Disentangling contributions of point and line defects in the Raman spectra of graphene-related materials. 2D Mater. 2017, 4, 025039. [Google Scholar] [CrossRef]
- Cuesta, A.; Dhamelincourt, P.; Laureyns, J.; Martínez-Alonso, A.; Tascón, J.M.D. Raman microprobe studies on carbon materials. Carbon 1994, 32, 1523–1532. [Google Scholar] [CrossRef]
- Beyssac, O.; Goffé, B.; Petitet, J.-P.; Froigneux, E.; Moreau, M.; Rouzaud, J.-N. On the characterization of disordered and heterogeneous carbonaceous materials by Raman spectroscopy. Georaman 2002, Fifth International Conference on Raman Spectroscopy. Appl. Earth Sci. 2003, 59, 2267–2276. [Google Scholar]
- Couzi, M.; Bruneel, J.-L.; Talaga, D.; Bokobza, L. A multi wavelength Raman scattering study of defective graphitic carbon materials: The first order Raman spectra revisited. Carbon 2016, 107, 388–394. [Google Scholar] [CrossRef]
- Berhanu, S.; Hervy, M.; Weiss-Hortala, E.; Proudhon, H.; Berger, M.-H.; Chesnaud, A.; Faessel, M.; King, A.; Pham Minh, D.; Villot, A.; et al. Advanced characterization unravels the structure and reactivity of wood-based chars. J. Anal. Appl. Pyrolysis 2018, 130, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Harris, P.J.F.; Tsang, S.C. High-resolution electron microscopy studies of non-graphitizing carbons. Philos. Mag. A 1997, 76, 667–677. [Google Scholar] [CrossRef]
- Singh, M.; Vander Wal, R.L. Nanostructure Quantification of Carbon Blacks. C 2019, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Monthioux, M.; Noé, L.; Kobylko, M.; Wang, Y.; Cazares-Huerta, T.C.; Pénicaud, A. Determining the structure of graphene-based flakes from their morphotype. Carbon 2017, 115, 128–133. [Google Scholar] [CrossRef]
- Lutterotti, L.; Scardi, P. Simultaneous structure and size–strain refinement by the Rietveld method. J. Appl. Crystallogr. 1990, 23, 246–252. [Google Scholar] [CrossRef]
- Neverov, V.S. XaNSoNS: GPU-accelerated simulator of diffraction patterns of nanoparticles. SoftwareX 2017, 6, 63–68. [Google Scholar] [CrossRef]
- Mallet-Ladeira, P.; Puech, P.; Weisbecker, P.; Vignoles, G.L.; Monthioux, M. Behavior of Raman D band for pyrocarbons with crystallite size in the 2–5 nm range. Appl. Phys. A 2014, 114, 759–763. [Google Scholar] [CrossRef]
- Puech, P.; Plewa, J.-M.; Mallet-Ladeira, P.; Monthioux, M. Spatial confinement model applied to phonons in disordered graphene-based carbons. Carbon 2016, 105, 275–281. [Google Scholar] [CrossRef]
- Cançado, L.G.; Jorio, A.; Pimenta, M.A. Measuring the absolute Raman cross section of nanographites as a function of laser energy and crystallite size. Phys. Rev. B 2007, 76, 064304. [Google Scholar] [CrossRef]
- Asadullah, M.; Zhang, S.; Min, Z.; Yimsiri, P.; Li, C.-Z. Effects of biomass char structure on its gasification reactivity. Bioresour. Technol. 2010, 101, 7935–7943. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hayashi, J.I.; Li, C.Z. FT-Raman spectroscopic study of the evolution of char structure during the pyrolysis of a Victorian brown coal. Fuel 2006, 85, 1700–1707. [Google Scholar] [CrossRef]
- Kang, D.S.; Lee, S.M.; Lee, S.H.; Roh, J.S. X-ray diffraction analysis of the crystallinity of phenolic resin-derived carbon as a function of the heating rate during the carbonization process. Carbon Lett. 2018, 27, 108–111. [Google Scholar]
- Kercher, A.; Nagle, D. Microstructural Evolution during Charcoal Carbonization by X-ray Diffraction Analysis. Carbon 2003, 41, 15–27. [Google Scholar] [CrossRef]
- Keiluweit, M.; Nico, P.S.; Johnson, M.G.; Kleber, M. Dynamic Molecular Structure of Plant Biomass-Derived Black Carbon (Biochar). Environ. Sci. Technol. 2010, 44, 1247–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahbi, M.; Kiso, M.; Kubota, K.; Horiba, T.; Chafik, T.; Hida, K.; Matsuyama, T.; Komaba, S. Synthesis of hard carbon from argan shells for Na-ion batteries. J. Mater. Chem. A 2017, 5, 9917–9928. [Google Scholar] [CrossRef]
- Campos-Delgado, J.; Kim, Y.A.; Hayashi, T.; Morelos-Gómez, A.; Hofmann, M.; Muramatsu, H.; Endo, M.; Terrones, H.; Shull, R.D.; Dresselhaus, M.S.; et al. Thermal stability studies of CVD-grown graphene nanoribbons: Defect annealing and loop formation. Chem. Phys. Lett. 2009, 469, 177–182. [Google Scholar] [CrossRef]
- Garvey, C.J.; Parker, I.H.; Simon, G.P. On the Interpretation of X-ray Diffraction Powder Patterns in Terms of the Nanostructure of Cellulose I Fibres. Macromol. Chem. Phys. 2005, 206, 1568–1575. [Google Scholar] [CrossRef]
- Qiao, Q.; Li, X.; Huang, L. Crystalline Cellulose under Pyrolysis Conditions: The Structure–Property Evolution via Reactive Molecular Dynamics Simulations. J. Chem. Eng. Data 2020, 65, 360–372. [Google Scholar] [CrossRef]
- Kwon, S.-M.; Kim, N.; Cha, D.-S. An investigation on the transition characteristics of the wood cell walls during carbonization. Wood Sci. Technol. 2009, 43, 487–498. [Google Scholar] [CrossRef]
- Meng, Y.; Contescu, C.; Liu, P.; Wang, S.; Lee, S.H.; Guo, J.; Young, T. Understanding the local structure of disordered carbons from cellulose and lignin. Wood Sci. Technol. 2021, 55, 587–606. [Google Scholar] [CrossRef]
- Noda, T.; Inagaki, M. The Structure of Glassy Carbon. Bull. Chem. Soc. Jpn. 1964, 37, 1534–1538. [Google Scholar] [CrossRef]
- Paris, O.; Zollfrank, C.; Zickler, G.A. Decomposition and carbonisation of wood biopolymers—A microstructural study of softwood pyrolysis. Carbon 2005, 43, 53–66. [Google Scholar] [CrossRef]
- Wu, H.; Gakhar, R.; Chen, A.; Lam, S.; Marshall, C.P.; Scarlat, R. Comparative analysis of microstructure and reactive sites for nuclear graphite IG-110 and graphite matrix A3. J. Nucl. Mater. 2019, 528, 151802. [Google Scholar] [CrossRef]
- Monthioux, M.; Charlier, J.-C. Giving credit where credit is due: The Stone–(Thrower)–Wales designation revisited. Carbon 2014, 75, 1–4. [Google Scholar] [CrossRef]
- McDonald-Wharry, J.; Manley-Harris, M.; Pickering, K.L. Carbonisation of biomass-derived chars and the thermal reduction of a graphene oxide sample studied using Raman spectroscopy. Carbon 2013, 59, 383–405. [Google Scholar] [CrossRef] [Green Version]
- Ouzilleau, P.; Gheribi, A.E.; Chartrand, P.; Soucy, G.; Monthioux, M. Why some carbons may or may not graphitize? The point of view of thermodynamics. Carbon 2019, 149, 419–435. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extracted Parameters | Significance |
|---|---|
| (ID/IG) | Calculation of La [19,22] |
| D band—FWHMD | Calculation of La [36] |
| D band | Degree of graphenic-sp2 disorder |
| Bands and Usual Position in Graphenic Materials (cm−1) | Fitting Function | Representation |
|---|---|---|
| G ≈ 1580 | Voigt | sp2 |
| D1 ≈ 1350 | Voigt | defects in sp2 (point defects, edges, curvature) |
| D2 ≈ 1620 | Gaussian | defects in sp2, needed due to fixed G band wavenumber and thus associated to disorder |
| D3 ≈ 1500 | Gaussian | amorphous carbon |
| Extracted Parameters | Treatment Conditions | |
|---|---|---|
| 1000 °C, 2 °C/min | 1800 °C, 2 °C/min | |
| Lc(nm) from 001 | 1.04 | 1.26 |
| La (nm) from 10 | 1.85 | 3.22 |
| La (nm) from 11 | 1.76 | 2.85 |
| d001 (Å) | 3.75 * (3.68 **) | 3.62 * (3.55 **) |
| aC = C (Å) | 1.42 | 1.42 |
| 3.46 | 3.46 | |
| 0.44 | 0.54 | |
| 0.20 | 0.21 | |
| 0.44 | 0.39 | |
| 1.65 | 0.87 | |
| Crystallite Size (nm) | 1000 °C, 2 °C/min | 1800 °C, 2 °C/min |
|---|---|---|
| La [19] | - | 2.45 |
| La [22] | 1.42 | - |
| La [36] | <2.84 | 8.69 |
| Excitation Wavelength 532 nm | 1000 °C, 2 °C/min | 1800 °C, 2 °C/min |
|---|---|---|
| Intensities: | 0.173 | 0.055 |
| Integrated intensities: | 0.153 | 0.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mubari, P.K.; Beguerie, T.; Monthioux, M.; Weiss-Hortala, E.; Nzihou, A.; Puech, P. The X-ray, Raman and TEM Signatures of Cellulose-Derived Carbons Explained. C 2022, 8, 4. https://doi.org/10.3390/c8010004
Mubari PK, Beguerie T, Monthioux M, Weiss-Hortala E, Nzihou A, Puech P. The X-ray, Raman and TEM Signatures of Cellulose-Derived Carbons Explained. C. 2022; 8(1):4. https://doi.org/10.3390/c8010004
Chicago/Turabian StyleMubari, Petros Kasaira, Théotime Beguerie, Marc Monthioux, Elsa Weiss-Hortala, Ange Nzihou, and Pascal Puech. 2022. "The X-ray, Raman and TEM Signatures of Cellulose-Derived Carbons Explained" C 8, no. 1: 4. https://doi.org/10.3390/c8010004
APA StyleMubari, P. K., Beguerie, T., Monthioux, M., Weiss-Hortala, E., Nzihou, A., & Puech, P. (2022). The X-ray, Raman and TEM Signatures of Cellulose-Derived Carbons Explained. C, 8(1), 4. https://doi.org/10.3390/c8010004