
Construction of the Pseudomonas putida Strain with Low Motility and Reduced Biofilm Formation for Application in Fermentation

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, and Growth Conditions
2.2. Construction of Recombinant Plasmids for Deletion of algA, pilQ, flhA Genes
2.3. Creating of the Mutant Strain P. putida PCL1760 with Knockout of the algA, pilQ and flhA Genes
2.4. Comparison of the Growth Rate of the P. putida LN6160 and PCL1760 Strains
2.5. Comparison of the Motility of P. putida PCL1760 Wild Type Strain and Its Mutants
2.6. Quantitative Comparison of P. putida PCL1760 Wild Type Strain and Its Mutants
2.7. Comparing Analysis of the Growth Rate of the Mutant and Wild-Type Strains in the Small-Scale Parallel Bioreactors
3. Results
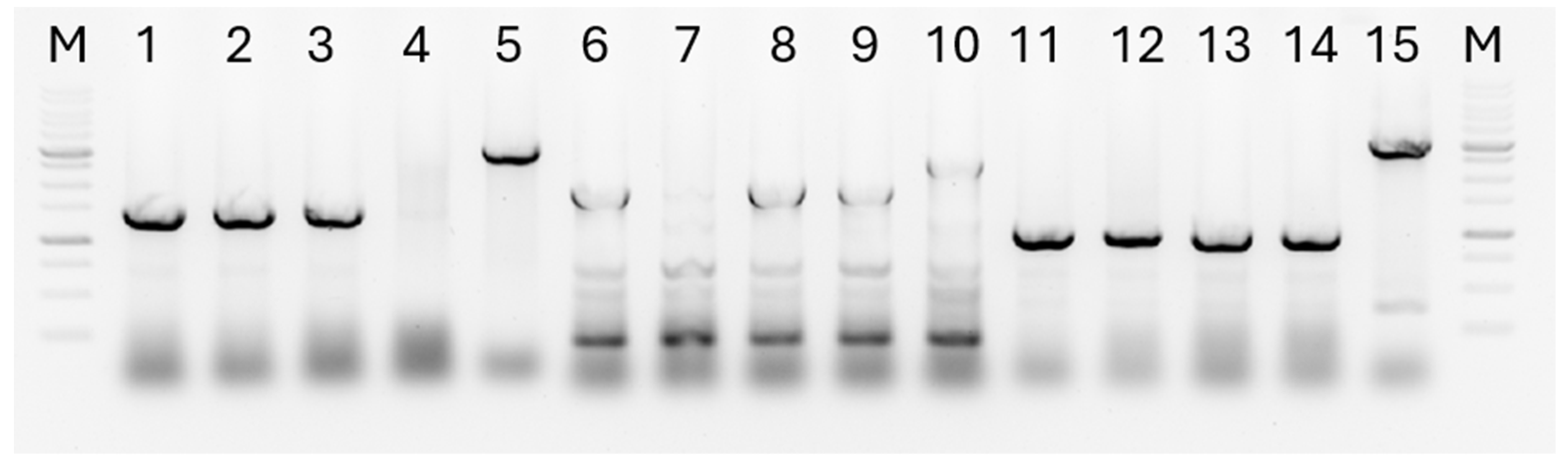
3.1. Construction of Recombinant Plasmids for Deletion of the algA, pilQ and flhA Genes
3.2. Construction of the P. putida LN6160 Strain
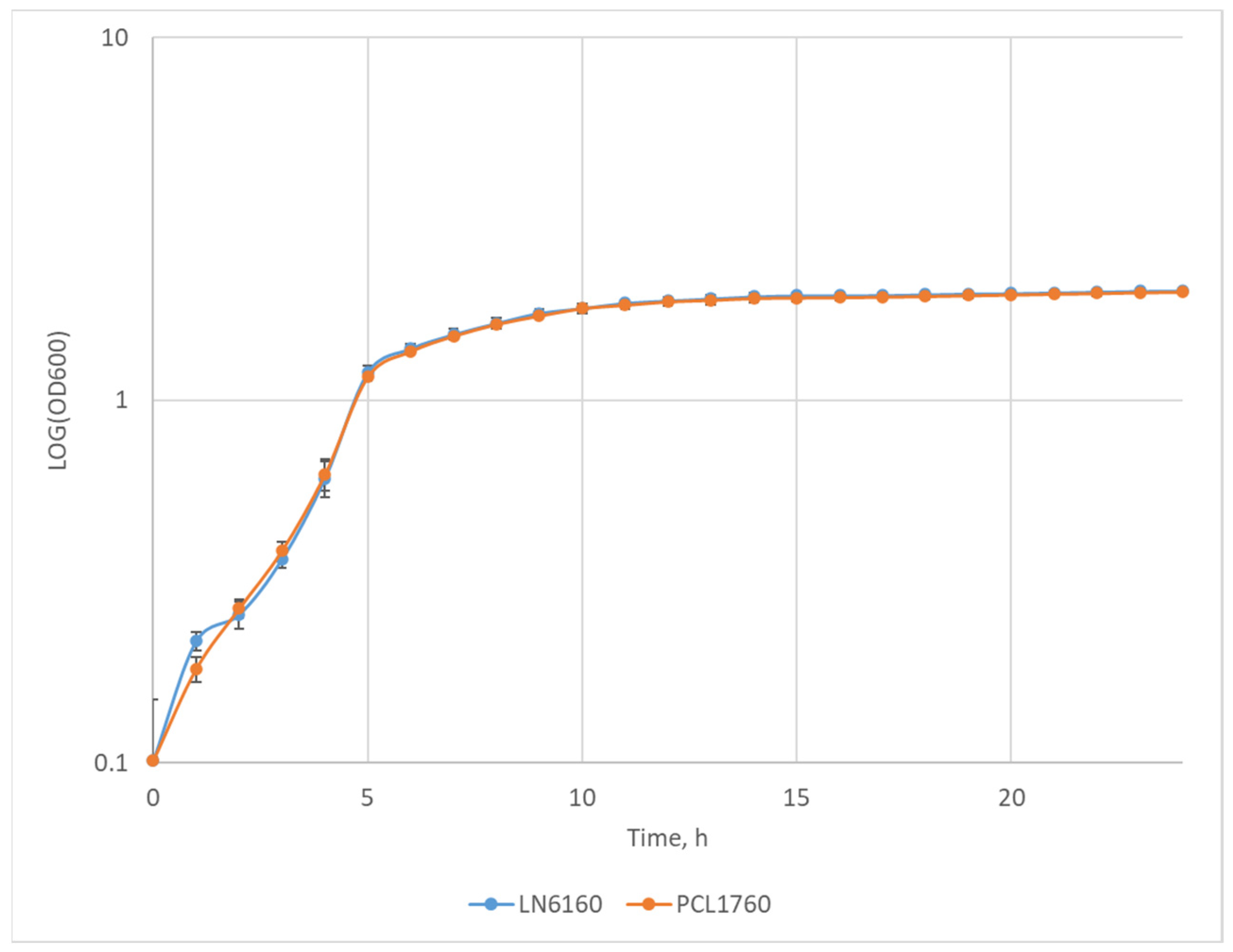
3.3. Comparison of the Growth Rate of the P. putida LN6160 and PCL1760 Strains
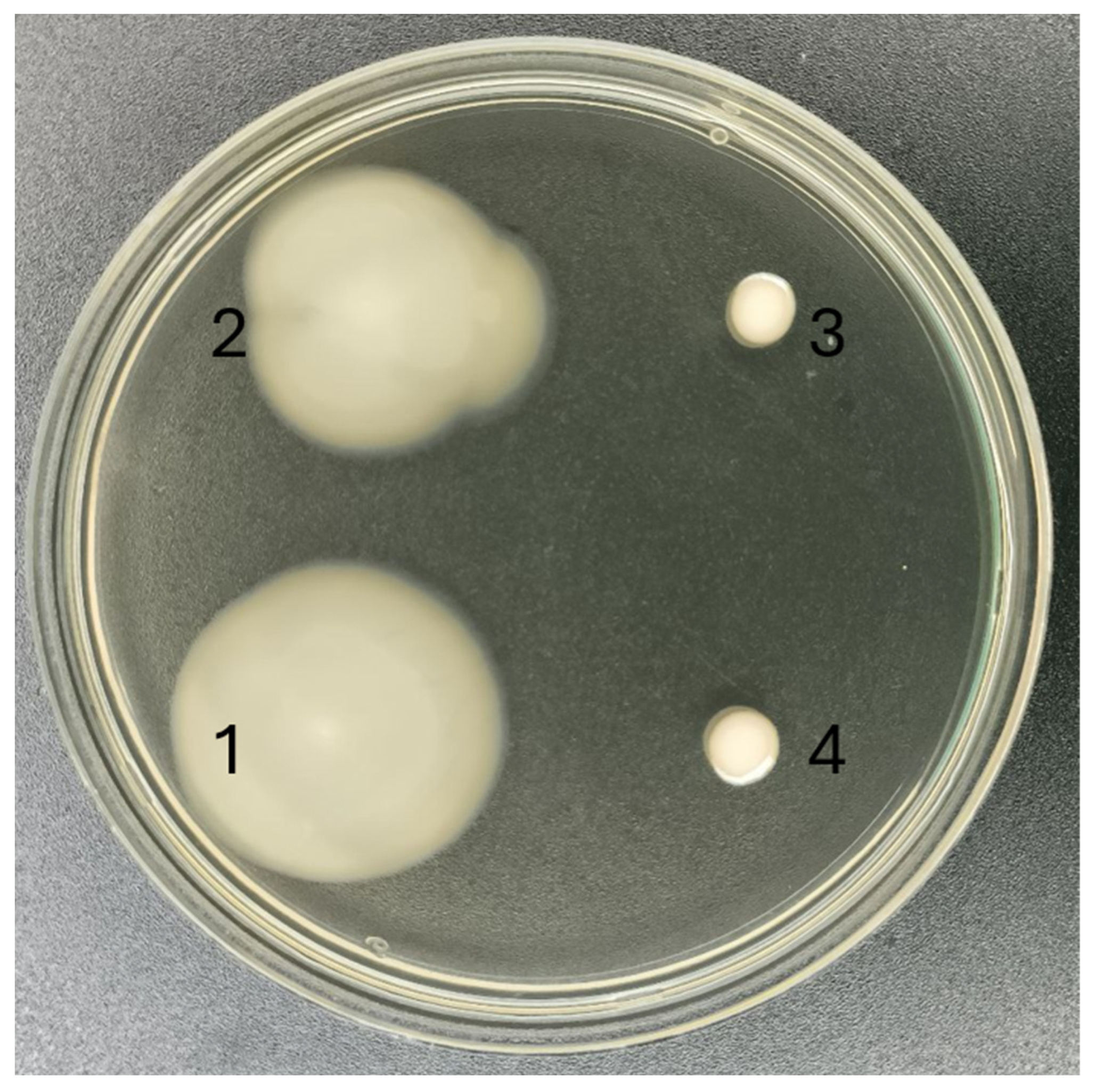
3.4. Motility of P. putida PCL1760 Wild Type Strain and Its Mutants
3.5. Quantitative Comparison of P. putida PCL1760 Wild Type Strain and Its Mutants Biofilm Formation
3.6. Comparative Cultivation of the LN6160 Mutant Strain and the Wild-Type LN6160 Strain in Small-Scale Bioreactors in Rich and Mineral Mediua
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Validov, S. Biological Control of Tomato Foot and Root Rot by Pseudomonas bacteria in Stonewool; Leiden University: Leiden, The Netherlands, 2007. [Google Scholar]
- Afordoanyi, D.M.; Diabankana, R.G.C.; Miftakhov, A.K.; Kuchaev, E.S.; Validov, S.Z. Genomic Features of Pseudomonas putida PCL1760: A Biocontrol Agent Acting via Competition for Nutrient and Niche. Appl. Microbiol. 2022, 2, 749–765. [Google Scholar] [CrossRef]
- Miftakhov, A.K.; Diabankana, R.G.C.; Frolov, M.; Yusupov, M.M.; Validov, S.Z.; Afordoanyi, D.M. Persistence as a Constituent of a Biocontrol Mechanism (Competition for Nutrients and Niches) in Pseudomonas putida PCL1760. Microorganisms 2023, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Koehorst, J.J.; van Dam, J.C.J.; van Heck, R.G.A.; Saccenti, E.; Martins dos Santos, V.A.P.; Suarez-Diez, M.; Schaap, P.J. Comparison of 432 Pseudomonas Strains through Integration of Genomic, Functional, Metabolic and Expression Data. Sci. Rep. 2016, 6, 38699. [Google Scholar] [CrossRef] [PubMed]
- Brehl, C.; Brass, H.U.C.; Lüchtrath, C.; Böckmann, L.; Ihling, N.; Classen, T.; Pietruszka, J.; Büchs, J. Optimized prodigiosin production with Pseudomonas putida KT2440 using parallelized noninvasive online monitoring. Biotechnol. Prog. 2022, 38, e3245. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.; Duane, G.; Kenny, S.T.; Cerrone, F.; Guzik, M.W.; Babu, R.P.; Casey, E.; O’Connor, K.E. High cell density cultivation of Pseudomonas putida KT2440 using glucose without the need for oxygen enriched air supply. Biotechnol. Bioeng. 2015, 112, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Escobar, S.; Gomez, E.; Santos, V.E.; Garcia-Ochoa, F. Behavior of several Pseudomonas putida strains growth under different agitation and oxygen supply conditions. Biotechnol. Prog. 2018, 34, 900–909. [Google Scholar] [CrossRef]
- Prabhukhot, G.S.; Eggleton, C.D.; Patel, J. Multispecies Bacterial Biofilms and Their Evaluation Using Bioreactors. Foods 2023, 12, 4495. [Google Scholar] [CrossRef]
- Yin, W.; Xu, S.; Wang, Y.; Zhang, Y.; Chou, S.-H.; Galperin, M.Y.; He, J. Ways to Control Harmful Biofilms: Prevention, Inhibition, and Eradication. Crit. Rev. Microbiol. 2021, 47, 57–78. [Google Scholar] [CrossRef]
- Hay, I.D.; Rehman, Z.U.; Moradali, M.F.; Wang, Y.; Rehm, B.H.A. Microbial alginate production, modification and its applications. Microb. Biotechnol. 2013, 6, 637. [Google Scholar] [CrossRef]
- Shinabarger, D.; Berry, A.; May, T.B.; Rothmel, R.; Fialho, A.; Chakrabarty, A.M. Purification and characterization of phosphomannose isomerase-guanosine diphospho-D-mannose pyrophosphorylase. A bifunctional enzyme in the alginate biosynthetic pathway of Pseudomonas aeruginosa. J. Biol. Chem. 1991, 266, 2080–2088. [Google Scholar] [CrossRef]
- Tatnell, P.J.; Russell, N.J.; Gacesa, P. GDP-mannose dehydrogenase is the key regulatory enzyme in alginate biosynthesis in Pseudomonas aeruginosa: Evidence from metabolite studies. Microbiology 1994, 140, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Haiko, J.; Westerlund-Wikström, B. The Role of the Bacterial Flagellum in Adhesion and Virulence. Biology 2013, 2, 1242. [Google Scholar] [CrossRef]
- Feldman, M.; Bryan, R.; Rajan, S.; Scheffler, L.; Brunnert, S.; Tang, H.; Prince, A. Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect. Immun. 1998, 66, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Bouteiller, M.; Dupont, C.; Bourigault, Y.; Latour, X.; Barbey, C.; Konto-ghiorghi, Y.; Merieau, A. Pseudomonas Flagella: Generalities and Specificities. Int. J. Mol. Sci. 2021, 22, 3337. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Ochman, H. Origins of Flagellar Gene Operons and Secondary Flagellar Systems. J. Bacteriol. 2007, 189, 7098. [Google Scholar] [CrossRef] [PubMed]
- Osterman, I.A.; Dikhtyar, Y.Y.; Bogdanov, A.A.; Dontsova, O.A.; Sergiev, P.V. Regulation of flagellar gene expression in Bacteria. Biochemistry 2015, 80, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Terashima, H.; Homma, M. Regulation of the Single Polar Flagellar Biogenesis. Biomolecules 2020, 10, 533. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Inoue, Y.; Kinoshita, M.; Namba, K. FLIK-driven conformational rearrangements of FlhA and FlhB are required for export switching of the flagellar protein export apparatus. J. Bacteriol. 2020, 202, e00637-19. [Google Scholar] [CrossRef] [PubMed]
- Terashima, H.; Kawamoto, A.; Tatsumi, C.; Namba, K.; Minamino, T.; Imada, K. In vitro reconstitution of functional type III protein export and insights into flagellar assembly. Mbio 2018, 9, e00988-18. [Google Scholar] [CrossRef]
- Bradley, D.E. Basic Characterization of a Pseudomonas aeruginosa Pilus-Dependent Bacteriophage with a Long Noncontractile. Tail. J. Virol. 1973, 12, 1139. [Google Scholar] [CrossRef]
- Tammam, S.; Sampaleanu, L.M.; Koo, J.; Manoharan, K.; Daubaras, M.; Burrows, L.L.; Howell, P.L. PilMNOPQ from the Pseudomonas aeruginosa Type IV Pilus System Form a Transenvelope Protein Interaction Network That Interacts with PilA. J. Bacteriol. 2013, 195, 2126. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Tammam, S.; Rubinstein, J.L.; Burrows, L.L.; Howell, P.L. CryoEM map of Pseudomonas aeruginosa PilQ enables structural characterization of TsaP. Structure 2021, 29, 457–466.e4. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Zhu, J.; Feng, L.; Li, J.; Liu, X. Characterization of LuxI/LuxR and Their Regulation Involved in Biofilm Formation and Stress Resistance in Fish Spoilers Pseudomonas fluorescens. Int. J. Food Microbiol. 2019, 297, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Norrander, J.; Kempe, T.; Messing, J. Construction of improved M13 vectors using oligodeoxynucleotide-directed mutagenesis. Gene 1983, 26, 101–106. [Google Scholar] [CrossRef]
- Simon, R.; Priefer, U.; Pühler, A. A Broad Host Range Mobilization System for In Vivo Genetic Engineering: Transposon Mutagenesis in Gram Negative Bacteria. Nat. Biotechnol. 1983, 1, 784–791. [Google Scholar] [CrossRef]
- Kvitko, B.H.; Collmer, A. Construction of Pseudomonas syringae pv. tomato DC3000 mutant and polymutant strains. Methods Mol. Biol. 2011, 712, 109–128. [Google Scholar] [CrossRef]
- Ha, D.G.; Kuchma, S.L.; O’Toole, G.A. Plate-based assay for swimming motility in Pseudomonas aeruginosa. Methods Mol. Biol. 2014, 1149, 59–65. [Google Scholar] [CrossRef]
- Stepanović, S.; Vuković, D.; Dakić, I.; Savić, B.; Švabić-Vlahović, M. A modified microtiter-plate test for quantification of staphylococcal biofilm formation. J. Microbiol. Methods 2000, 40, 175–179. [Google Scholar] [CrossRef]
- Loeschcke, A.; Thies, S. Pseudomonas putida-a versatile host for the production of natural products. Appl. Microbiol Biotechnol. 2015, 99, 6197. [Google Scholar] [CrossRef]
- Nikel, P.I.; de Lorenzo, V. Pseudomonas putida as a functional chassis for industrial biocatalysis: From native biochemistry to trans-metabolism. Metab. Eng. 2018, 50, 142–155. [Google Scholar] [CrossRef]
- Weimer, A.; Kohlstedt, M.; Volke, D.C.; Nikel, P.I.; Wittmann, C. Industrial biotechnology of Pseudomonas putida: Advances and prospects. Appl. Microbiol. Biotechnol. 2020, 104, 7745–7766. [Google Scholar] [CrossRef] [PubMed]
- de Lorenzo, V.; Pérez-Pantoja, D.; Nikel, P.I. Pseudomonas putida KT2440: The long journey of a soil-dweller to become a synthetic biology chassis. J. Bacteriol. 2024, 206, e0013624. [Google Scholar] [CrossRef]
- Validov, S.Z.; Kamilova, F.; Lugtenberg, B.J.J. Pseudomonas putida strain PCL1760 controls tomato foot and root rot in stonewool under industrial conditions in a certified greenhouse. Biol. Contr. 2009, 48, 6–11. [Google Scholar] [CrossRef]
- Fontaine, F.; Stewart, E.J.; Lindner, A.B.; Taddei, F. Mutations in two global regulators lower individual mortality in Escherichia coli. Mol. Microbiol. 2008, 67, 2–14. [Google Scholar] [CrossRef]
- Ma, L.; Conover, M.; Lu, H.; Parsek, M.R.; Bayles, K.; Wozniak, D.J. Assembly and development of the Pseudomonas aeruginosa biofilm matrix. PLoS Pathog. 2009, 5, e1000354. [Google Scholar] [CrossRef]
- Colvin, K.M.; Alnabelseya, N.; Baker, P.; Whitney, J.C.; Howell, P.L.; Parsek, M.R. PelA deacetylase activity is required for Pel polysaccharide synthesis in Pseudomonas aeruginosa. J. Bacteriol. 2013, 195, 2329–2339. [Google Scholar] [CrossRef]
- Qi, Y.H.; Huang, L.; Liu, G.F.; Leng, M.; Lu, G.T. PilG and PilH antagonistically control flagellum-dependent and pili-dependent motility in the phytopathogen Xanthomonas campestris pv. campestris. BMC Microbiol. 2020, 20, 37. [Google Scholar] [CrossRef]
- Ramadhan, S.H.; Matsui, T.; Nakano, K.; Minami, H. High cell density cultivation of Pseudomonas putida strain HKT554 and its application for optically active sulfoxide production. Appl. Microbiol. Biotechnol. 2013, 97, 1903–1907. [Google Scholar] [CrossRef]
- Chung, J.; Eisha, S.; Park, S.; Morris, A.J.; Martin, I. How Three Self-Secreted Biofilm Exopolysaccharides of Pseudomonas aeruginosa, Psl, Pel, and Alginate, Can Each Be Exploited for Antibiotic Adjuvant Effects in Cystic Fibrosis Lung Infection. Int. J. Mol. Sci. 2023, 24, 8709. [Google Scholar] [CrossRef]
- Friedman, L.; Kolter, R. Genes Involved in Matrix Formation in Pseudomonas aeruginosa PA14 Biofilms. Mol. Microbiol. 2004, 51, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Núñez, C.; López-Pliego, L.; Ahumada-Manuel, C.L.; Castañeda, M. Genetic Regulation of Alginate Production in Azotobacter vinelandii a Bacterium of Biotechnological Interest: A Mini-Review. Front. Microbiol. 2022, 13, 845473. [Google Scholar] [CrossRef] [PubMed]
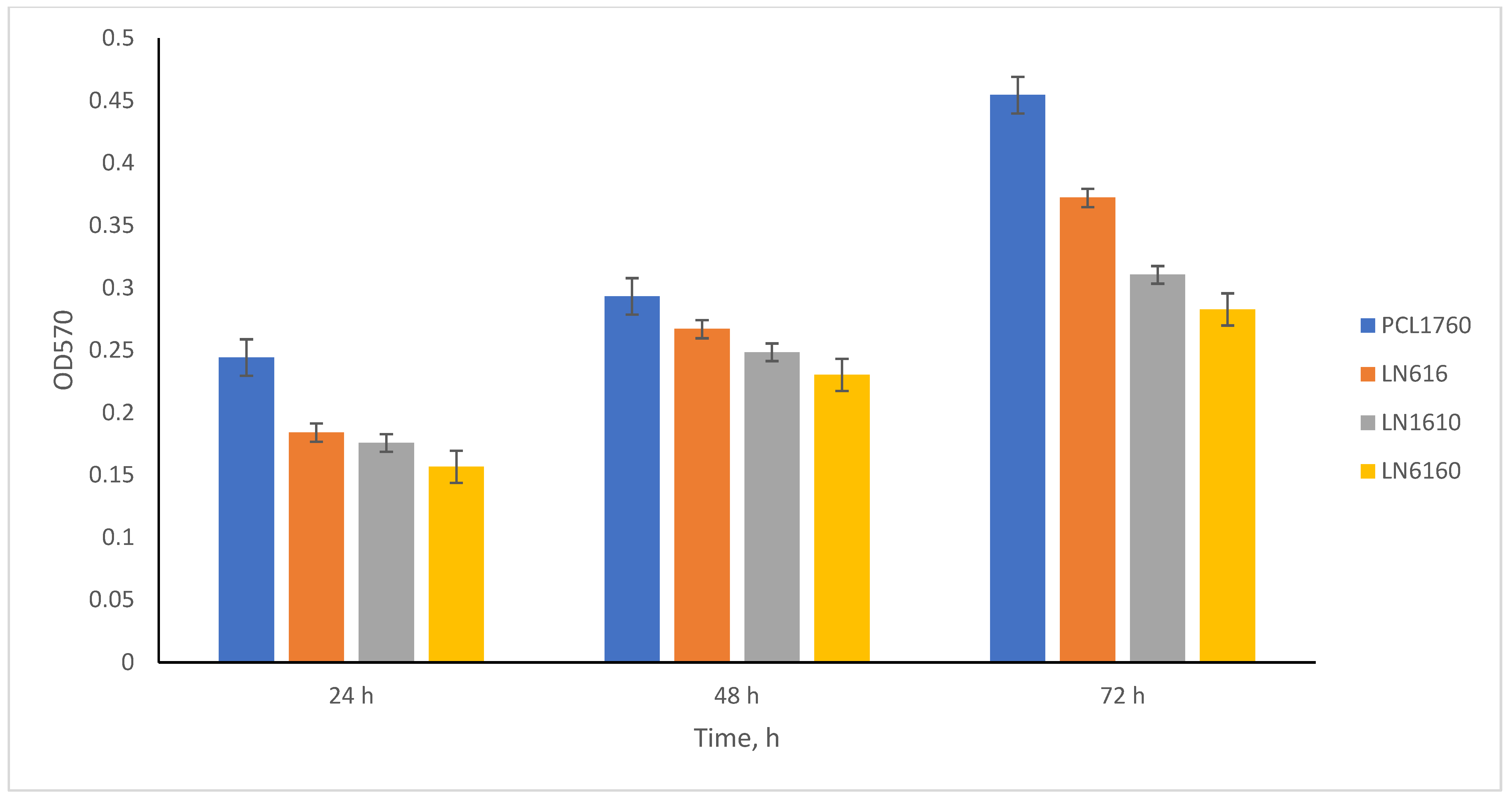


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain/Plasmid | Features | Reference/Source |
|---|---|---|
| E. coli DH5α | F-80ΔlacZ M15 (lacZYA-argF) U169 recA1 endA1hsdR17(rk−, mk+) phoA supE44-thi-1 gyrA96 relA1 | [25] |
| E. coli S17-1 | recA pro (RP4-2Tet: Mu) | [26] |
| P. putida PCL1760 | Wild type | Lab stock |
| P. putida LN616 | ΔalgA, | This work |
| P. putida LN1610 | ΔalgA, ΔflhA | This work |
| P. putida LN6160 | ΔalgA, ΔpilQ, ΔflhA | This work |
| pK18mobSacB | sacB lacZα Kmr; cloning vector for allelic exchange | [27] |
| pK18mobSacB-∆algA | pK18mobsacB carrying a modified algA gene region | This work |
| pK18mobSacB-∆pilQ | pK18mobsacB carrying a modified pilQ gene region | This work |
| pK18mobSacB-∆flhA | pK18mobsacB carrying a modified flhA gene region | This work |
| Primer | Nucleotide Sequence 5′-3′ |
|---|---|
| algA-fl1-for | CGAATTCGAGCTCGGTACCCACCTGGCCCTCGGTATCG |
| algA-fl1-rev | CCACCCACCACCCTGAAAGGTCGCTGCAAATC |
| algA-fl2-for | CCTTTCAGGGTGGTGGGTGGCCCATTCG |
| algA-fl2-rev | GTCGACTCTAGAGGATCCCCCCCGAACGTTATCTGCCAATGAAAAAC |
| test-algA-for | CTTGGCTGGGTAACGCAACAACAACAC |
| test-algA-rev | GGACAGACCACTGACTGATAGAGAGG |
| pilQ-fl1-for | CGAATTCGAGCTCGGTACCCGCTGCAAGCGCTCGAGAATATCG |
| pilQ-fl1-rev | ACCAGGGTGAAAAAAAGCGGCAACGGCGAGATC |
| pilQ-fl2-for | CCGCTTTTTTTCACCCTGGTAAGGCGTCGC |
| pilQ-fl2-rev | GTCGACTCTAGAGGATCCCCGCTCGCTTGGCTGGCGATG |
| test-pilQ-for | GGCTTGCTTGTCGAACGCCTCGATG |
| test-pilQ-rev | CGGCGACCTGCGCGGTAACG |
| flhA-fl1-for | CGAATTCGAGCTCGGTACCCTGTTGCAGCCAAAATTCAGC |
| flhA-fl1-rev | ATCCCCTACCCGCGAGTCCTCTTGATGC |
| flhA-fl2-for | AGGACTCGCGGGTAGGGGATAATGCAAGTTAAGC |
| flhA-fl2-rev | GTCGACTCTAGAGGATCCCCAGGTAGTTCTTCGCGGCAATG |
| test-flhA-for | GCAACCCGACGCATTATGCGGTGG |
| test-flhA-rev | GCCAGCGAACCGAGTTGCACTTCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frolov, M.; Kungurov, G.A.; Valiakhmetov, E.E.; Gogov, A.S.; Trachtmann, N.V.; Validov, S.Z. Construction of the Pseudomonas putida Strain with Low Motility and Reduced Biofilm Formation for Application in Fermentation. Fermentation 2024, 10, 606. https://doi.org/10.3390/fermentation10120606
Frolov M, Kungurov GA, Valiakhmetov EE, Gogov AS, Trachtmann NV, Validov SZ. Construction of the Pseudomonas putida Strain with Low Motility and Reduced Biofilm Formation for Application in Fermentation. Fermentation. 2024; 10(12):606. https://doi.org/10.3390/fermentation10120606
Chicago/Turabian StyleFrolov, Mikhail, Galim Alimzhanovich Kungurov, Emil Elmirovich Valiakhmetov, Artur Sergeyevich Gogov, Natalia Viktorovna Trachtmann, and Shamil Zavdatovich Validov. 2024. "Construction of the Pseudomonas putida Strain with Low Motility and Reduced Biofilm Formation for Application in Fermentation" Fermentation 10, no. 12: 606. https://doi.org/10.3390/fermentation10120606
APA StyleFrolov, M., Kungurov, G. A., Valiakhmetov, E. E., Gogov, A. S., Trachtmann, N. V., & Validov, S. Z. (2024). Construction of the Pseudomonas putida Strain with Low Motility and Reduced Biofilm Formation for Application in Fermentation. Fermentation, 10(12), 606. https://doi.org/10.3390/fermentation10120606