1. Introduction
Enzymes as biocatalysts are very efficient, specific, and accelerate the reaction by more than 1-million-fold as compared to an uncatalyzed reaction [
1]. Microbes are a source of a wide variety of enzymes that have various industrial and therapeutic applications as biocatalysts. The advent of metagenomics, genome mining, and exploration studies of extremophiles have aided in exploring the microbial enzyme diversity. Due to their easy susceptibility to genetic manipulation, microbes are also used for recombinant expression of enzymes with high yield and minimal downstream purification process [
2].
Serratiopeptidase (also known as serralysin/serrapeptase/serratia protease) is a proteolytic enzyme originally reported to be produced by
Serratia marcescens. The enzyme also finds therapeutic application as an anti-inflammatory, anti-edemic, analgesic and fibrinolytic agent [
3]. Positive effects of serratiopeptidase have been reported in different medical procedures including orthopaedics, ophthalmology, surgery, gynaecology, otorhinolaryngology, pulmonology, and dentistry [
4]. Studies on rat models have also shown that in Alzheimer’s disease, serratiopeptidase may also be considered as a neuroprotective agent since it alleviates the neuroinflammation and apoptosis associated with the disease [
5]. Moreover, due to its mucolytic property, serratiopeptidase has also been proposed to act as a potent drug in the integrative management of COVID-19 [
6].
The current demand of serratiopeptidase in pharmaceuticals and industries is met by the use of a wild or mutated form of its natural strain
S. marcescens. The pathogenic nature of the producer bacteria and the threats associated with the disposal of the bulk biomass after fermentation demand a need to look into alternative production strategies using recombinant DNA technology. Reports for recombinant production of serratiopeptidase are available but many of them could not produce an active enzyme due to unstable expression plasmids, the formation of inclusion bodies, or the production of inactive enzyme [
7]. Successful expression and refolding of recombinant serratiopeptidase from inclusion bodies in different
E. coli vectors was reported in some recent studies [
8,
9]. However, high-yield and cost-effective production of recombinant serratiopeptidase by bioreactor level fermentation has not yet been reported.
Although shake flask is a standard tool in microbiology laboratory for culturing microbes, commercial production of any microbial product demands bioreactor-level production studies since several parameters crucial to culture performance, which can be monitored and controlled in the bioreactor, cannot be done at the flask level.
After successful clone selection and screening for recombinant protein production at the flask level, bioreactor systems are used for the large-scale production to fulfill the demands of industrial biopharmaceuticals using high throughput process development techniques [
10]. Even though a significant amount of literature is available for fed batch production of recombinant proteins in
E. coli [
11,
12,
13], optimum conditions for culture growth, product formation, and accumulation need to be defined for each expression system due to variations in promoters, sequence of expressed recombinant protein, and its effect on host physiology. Earlier reports [
13,
14] also stated that even though parameters like plasmid stability, copy number, as well as expressed product stability and solubility, are defined by the genes of host and vectors, the culture conditions and media composition also affect the same. Hence, each system needs optimization in terms of culture conditions and media constituents to achieve maximum product accumulation.
The current study presents optimization for high-density fed batch fermentation process for recombinant serratiopeptidase production. Using the statistically optimized media (OM) reported in earlier study [
9] as basal media, both batch and fed-batch fermentations were performed for maximizing the yield in terms of cell mass as well as recombinant protein product. Optimization for efficient lysis and solubilization of inclusion bodies from the bulk pellet followed by on-column refolding was done to obtain active serratiopeptidase from inclusion bodies.
2. Materials and Methods
2.1. Bacterial Strain and Expression Vector
For expression of the recombinant protein, the Champion™ pET SUMO Expression System (Invitrogen life sciences) in
E. coli BL21 (DE3) bacterial strain was used. The cloning procedure was described in our earlier study [
9]. Briefly, the serratiopeptidase gene was amplified using primers designed from the sequences already available in NCBI followed by TA cloning and expression studies of the clone having proper gene orientation. The 16s r-RNA sequence of the
Serratia marcescens strain (GenBank accession number MN945437) used as a source of serratiopeptidase gene and complete ORF of the cloned gene (GenBank accession number MN967013) is already deposited in NCBI.
2.2. Basal Media Components and Chemicals
All chemicals required for cell culture were purchased from Himedia. Luria Bertani (LB) broth was used as seed culture media; the OM was used as basal media for the bioreactor cultivation experiments. The OM contained Glucose (16 g L
−1), Soyapeptone (66 g L
−1), dipotassium hydrogen phosphate (12.7 g L
−1), magnesium sulfate (1.2 g L
−1), and trace metal solution (25 mL L
−1). The composition of the trace metal solution is given in
Table 1.
Feed media for obtaining high-density cell mass contained a carbon source (glycerol and/or glucose) and nitrogen source (Yeast extract and peptone), trace metal solution, and magnesium sulphate. Glucose and trace metal solution were autoclaved separately and the media was aseptically reconstituted for both basal and feed media. Kanamycin was filter sterilized and added at a final concentration of 100 mg L−1. Further, 50 mg L−1 of kanamycin sulfate was supplemented every 5 h to maintain the selection pressure. Expression of the protein was induced with 1 mM of filter sterilized isopropyl β-D-1- thiogalactopyranoside (IPTG).
2.3. Inoculum Preparation
A loopful of culture from frozen glycerol stock of the clone was revived on LB agar plate containing 50 mg L−1 kanamycin sulfate. A single colony from this plate was grown overnight at 37 °C in LB broth containing 50 mg L−1 kanamycin sulfate. This overnight grown culture was used as an inoculum (5% v/v) for the bioreactor culture.
2.4. Bioreactor Cultivation
All the bioreactor experiments were performed in a 5 L New Brunswick BioFlo 120 reactor (Eppendorf, Hamburg, Germany) at pH 7.0 and 30 °C temperature. 3 N Hydrochloric acid (HCl) and 3 N Sodium hydroxide (NaOH) solutions were used to maintain the pH. The dissolved oxygen (DO) concentration was monitored as a percentage of air saturation by using a polarographic, autoclavable oxygen electrode (Mettler Toledo). A constant aeration of 1 vessel volume per minute (vvm) was used during the batches. Cascading mode along with agitation and aeration was used to cope with dissolved oxygen concentration (setpoint 50%) by increasing the agitation rate. Different parameters like impeller speed, temperature, pH, and DO saturation (%) during the batch were continuously monitored online with the help of the fermenter software. The culture media was sampled every hour and the OD600 reading was taken to monitor culture growth. For determining the segregation stability of the plasmid, parallel plating of diluted sample aliquots was done on LB plates (Kan+ and Kan−), and colony-forming units of plasmid bearing cells was determined out of the total number of viable cells. At the end of the cultivation period, a small volume of the culture was sampled to check the protein expression on SDS-PAGE.
2.4.1. Batch Mode
Experiments in batch mode were performed to compare the yield in LB broth and OM. The culture was induced when the OD600 reached 3. The batch was harvested when an increase in the level of DO was observed along with a decrease in impeller speed.
2.4.2. Fed Batch Mode
Fed batch experiments were also started as a batch using OM. The composition of the feed media comprised carbon source (17.5% v/v of basal media), nitrogen source (5% w/v of basal media), and trace metal solution (0.625% v/v of basal media). Like batch mode, cascading along with aeration was used to provide the DO for the high-density fermentation. Feeding was initiated at the rate of 2.5% v/v of basal media per hour upon exhaustion of nutrients as indicated by a decrease in impeller speed and increase in DO concentration, and continued at the same rate. Induction was given after supplementation of 60% of the feed media volume. The batch was harvested when a sharp increase in DO concentration was observed.
The fed batch experiments were carried out to study the effect of carbon sources in feed media viz. glycerol and glucose. To augment the dissolved oxygen requirement of the cultures during the batches, cascade mode was used at different agitation (300 to 800 rpm or 300 to 1200 rpm) and aeration rates (1 vvm to 2.5 vvm). The yields in terms of cell mass and induced protein were determined.
2.5. Purification of Recombinant Serratiopeptidase
2.5.1. Optimization of Cell Lysis
The recombinant serratiopeptidase was found to be expressed in the form of inclusion bodies. Protein purification from inclusion bodies in bulk pellet following cell lysis prior to solubilization of inclusion bodies facilitates the removal of unnecessary soluble cytoplasmic proteins. The cell lysis process was optimized with the use of minimum steps for obtaining partially purified inclusion bodies. Cell lysis was performed by sonication. For this, the induced cell pellet was resuspended in 10× (w/v) volume of the basal buffer (20 mM Tris, 100 mM NaCl, pH 8) and sonicated in bath sonicator at high amplitude using 30s ON and 30s OFF cycles for 10 min on ice. Nucleic acids were removed by Benzonase treatment (1 U mL−1) for 30 min at 37 °C on a shaker. The lysis efficiency and induced protein loss by sonication were checked by performing SDS PAGE of the pellet and soluble fraction after sonication.
2.5.2. Optimization of Solubilization of Inclusion Bodies
Solubilization optimization of recombinant serratiopeptidase from inclusion bodies was performed using different methods including varying urea concentration (2–7 M) in the solubilization buffer, varying the pH of the solubilization buffer (pH 6 and 8–12), use of detergent, and freeze thaw method. The optimum solubilization procedure was determined by performing SDS PAGE of the solubilized and pellet fractions after solubilization. For the purification experiments, the method showing the presence of the maximum induced protein fraction in the solubilized fraction and involving the use of minimum resources was adopted. A wash of half volume of the solubilization buffer was given to solubilize any remaining inclusion bodies in the pellet fraction after solubilization.
2.5.3. Ni NTA Affinity Purification and on Column Refolding of Recombinant Serratiopeptidase
The recombinant serratiopeptidase was purified by affinity chromatography using NuviaTM IMAC resin (BioRad). The column was equilibrated with three column volumes (CV) of solubilization buffer, followed by the addition of solubilized inclusion bodies for binding of the recombinant protein with a subsequent wash of 3 CV of solubilization buffer. Column refolding was performed by giving the next wash with 10 CV of buffer containing 20 mM Tris (pH 8), 100 mM NaCl, and 0.1% triton-x 100, followed by wash with 10 CV of the same buffer without triton-x 100 to remove any residual detergent from the column. Elution was performed with native elution buffer containing 20 mM Tris (pH 8), 100 mM NaCl, and 250 mM imidazole. The purified enzyme was dialyzed against a buffer containing 10 mM Sodium acetate and 5 mM calcium, pH 7.5.
2.6. Determination of Specific Activity of Recombinant Serratiopeptidase
Caseinolytic assay [
15] was used to determine the specific activity of the serratiopeptidase enzyme with minor modifications. Casein substrate solution (0.65% in 50 mM potassium phosphate buffer pH 7.5) was used to check the proteolytic activity. Substrate solution (500 µL) was mixed with purified enzyme (100 µL) and incubated at 37 °C for 10 min. The reaction was stopped by the addition of 500 µL of 110 mM TCA followed by 30 min incubation at 37 °C. The reaction mixture was centrifuged at 10,000×
g for 10 min to pellet the precipitated protein and supernatant was filtered through a 0.45 µ filter. To 200 µL of supernatant, 500 µL of 500 mM sodium carbonate solution was added, followed by the addition of 100 µL Folin Ciocalteu (FC) reagent. The mixture was incubated at 37 °C for 30 µmin for the formation of a blue-colored chromophore by the reaction between tyrosine and FC reagent, and it was colorimetrically estimated by recording absorbance at 660 nm. The amount of tyrosine released was estimated by plotting a standard graph of known concentrations of tyrosine against its absorbance. One unit of proteolytic enzyme is defined as the amount of enzyme that will produce 1 µmole of tyrosine per minute. Enzyme units were calculated using the formula given below
2.7. Identification of Recombinant Protein by Mass Spectrometry
The identity of the pure protein was confirmed by peptide mass fingerprinting, wherein the band of pure protein was excised from the SDS-PAGE gel and subjected to in-gel trypsin digestion using mass spectrometry grade trypsin (Promega). The kit manufacturer’s protocol was followed for protein trypsinization, followed by purification and concentration of the digested fragments. The eluted peptides were subjected to mass spectrometric analysis using the Autoflex Speed MALDI-TOF/TOF mass spectrometer (Bruker Daltonics, Billerica, MA, USA). The peak data was submitted to MASCOT server for peptide mass fingerprinting analysis. Subsequently, one of the obtained peaks was fragmented for MS/MS analysis and the fragmented peak data was submitted on MASCOT server for MS/MS Ion search.
2.8. Intact Mass Analysis of Purified Recombinant Serratiopeptidase by LC-MS
Accurate mass of the purified auto-cleaved recombinant serratiopeptidase was determined by mass spectrometry to determine the exact cleavage site in the protein. Analysis was performed using AdvanceBio 6545XT Q-ToF (Agilent Technologies, Santa Clara, CA, USA). A PLRP-S reversed phase column (50 mm × 2.1 mm, 1000 A°) from Agilent was used for Liquid Chromatography system. Isocratic flow consisting of 50% Acetonitrile and 50% water was used at a flow rate of 0.3 mL min−1. The run time was 5 min. The flow was directed to the QToF mass analyzer through an ESI ion source. The nozzle voltage was set at 2000 V and the capillary voltage was set to 5500 V. Data was acquired using Agilent MassHunter Acquisition software and spectra deconvulation was done using the Agilent Masshunter Bioconfirm software.
3. Results and Discussion
3.1. Bioreactor Cultivation
Both batch and fed-batch fermentations were carried out to check the protein yield at the bioreactor level.
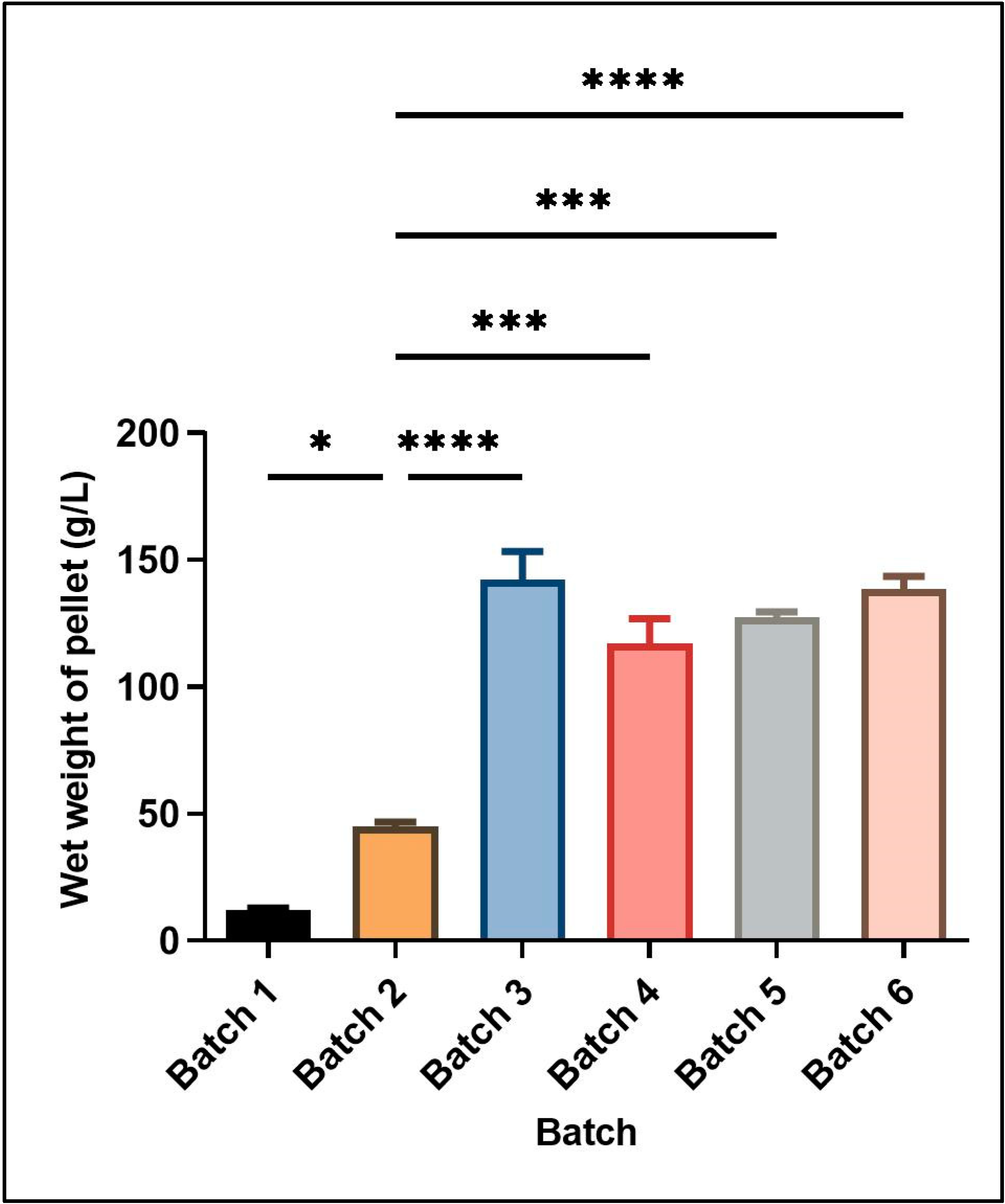
Figure 1 shows the wet cell mass obtained under different culture conditions in batch and fed-batch cultivations. The different culture conditions and relative yield of wet cell mass for each batch are mentioned in
Table 2.
Two different feeding strategies were attempted for enhancing cell mass and induced protein concentration consisting of the use of different carbon sources in the feed media at the rate of 2.5% v/v of the starting basal media volume. In the first strategy, only glycerol was used as a carbon source for feeding. The growth was continuously monitored till the time of harvest, yielding a wet cell mass of 142 ± 11.31 g L−1 media. In the second set of experiments, initial feeding was done using glucose containing feed media (7.5% w/v of basal media volume). Upon the exhaustion of nutrients, second feed supplementation containing glycerol (10% v/v of basal media volume) was started and induction was also given at the same time in this feed strategy, leading to a wet cell mass of 117 ± 9.9 g L−1. Thus, using glucose supplementation in feed media for recombinant serratiopeptidase production generated a smaller amount of cell mass.
Although glucose is a simpler carbon source readily used by most of the microbes, a higher cell mass and corresponding induced protein yield was obtained in this study using only glycerol in feed media as compared to glucose followed by glycerol. Similar results have been reported by Korz et al., 1995, where a higher biomass was obtained in glycerol as feed media as compared to glucose, mainly due to the higher concentration of glycerol in the feed medium since glucose has limited solubility [
16].
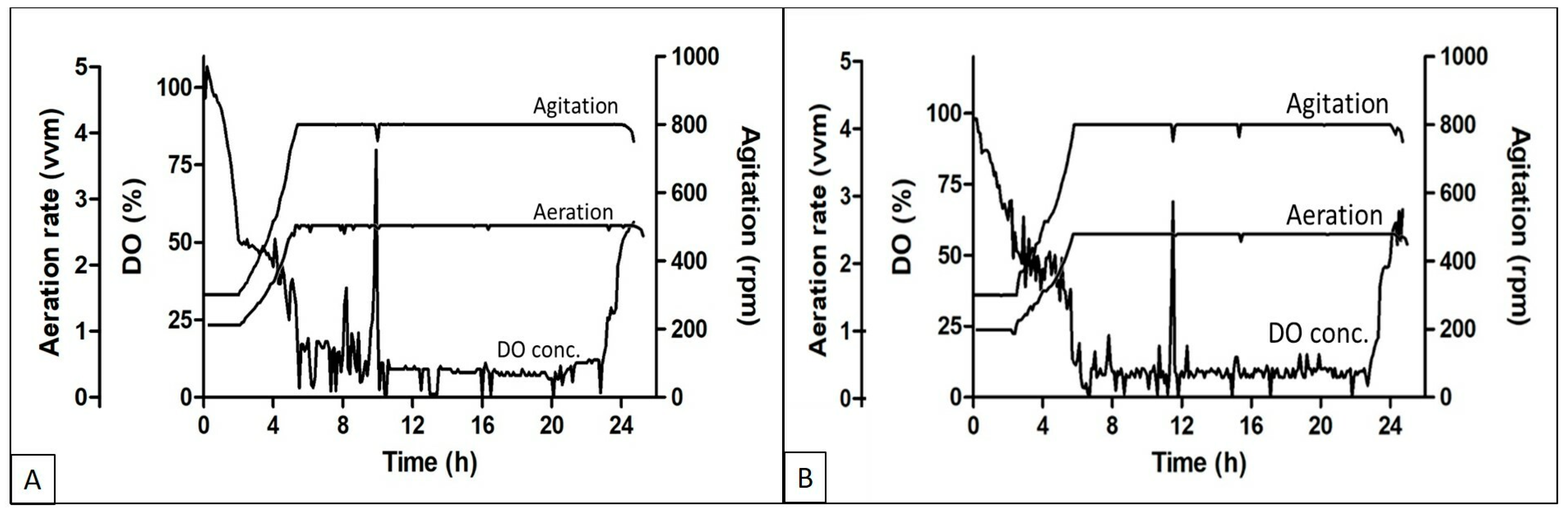
The DO, aeration, and agitation trend during the fed batch experiments using different feed media are shown in
Figure 2A,B respectively. The DO concentration started decreasing soon after the batch was started and reached the set threshold of 50% within 3–4 h of inoculation, followed by an increase in agitation and aeration rate to cope with the DO concentration in the media. The aeration and agitation rates were at their maximum set points till the exhaustion of the nutrients. Fed media supplementation was initiated when an increase in the DO concentration was observed. Soon after the feeding initiation, the DO went down and again the aeration and agitation reached the maximum set point. Although the batches were run in cascade mode along with aeration and agitation, the DO concentration was in the range of 5 to 15%.
Protein expression in high density fermentation is affected by the oxygen concentration in the medium at the time of growth. Oxygen is also known to affect both the cell density and recombinant protein formation during such processes. Lower oxygen concentrations lead to less production of respiratory ATP, which affects the activity of ATP-dependent chaperons that help in protein production and folding [
17].
In an attempt to further increase the reduced oxygen concentration during the batch, experiments were performed using the higher agitation rate (1200 rpm) to facilitate the mixing of air, leading to an increase in the dissolved oxygen concentration. The relative yield of cell mass and induced protein was found to be decreased at the higher agitation rate (Batch 5). No improvement was observed in the dissolved oxygen concentration at a higher agitation rate. The lower cell mass at higher agitation rate could be due to mechanical stress induced in the cells, leading to a reduction in the expression. Hence, further batches were performed using lower agitation rates. Studies have also reported that solubility and proper folding of the recombinant protein is enhanced at lower agitation rates [
18].
Batch 6 was performed using the above optimized conditions with a gradual reduction in the temperature to 20 °C after induction, since lower temperatures are also reported to enhance the protein over expression [
19,
20]. In our studies, the overall cell mass was found to be reduced at a lower temperature as compared to batch 3 where the incubation temperature was 30 °C. Also, the protein expression per unit of cell mass was also similar at both temperatures as confirmed by densitometry. Thus, the set of conditions used in batch 3 yielded maximum cell mass and induced protein expression.
Plasmid retention in cells is a significant parameter to be considered for high-density fed-batch fermentations since the gene for expression of protein of interest is present on the plasmid. The plasmid retention was confirmed by the presence of the plasmid-associated kanamycin resistance in the cells. It was observed that more than 99% of the cells retained the plasmid as checked at different time intervals during the batch. Plasmid stability studies were performed by spreading periodic culture dilutions on plates with and without kanamycin and estimating the colony-forming units (cfu / mL) in both the plates. There was no reduction in the number of colonies on the Kan+ plates as compared to Kan− plates, thus indicating no plasmid loss during the batches. Thus, the supplementation of antibiotic used during the batches was sufficient to maintain selection pressure during the long cultivation periods.
3.2. Purification of Recombinant Serratiopeptidase
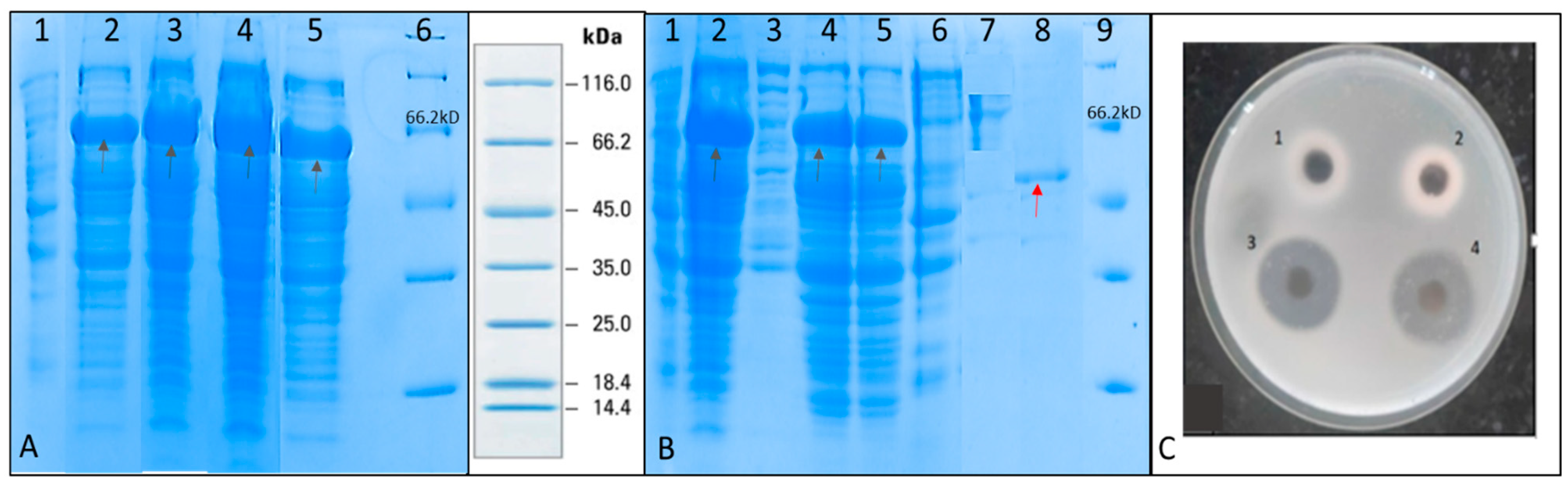
SDS PAGE was performed to check for the induction of recombinant serratiopeptidase in the different batches (
Figure 3A). It was observed that maximum-induced protein was obtained in fed batch containing glycerol feed media in the agitation range from 300–800 rpm. The purification of recombinant serratiopeptidase from induced pellet of this batch was used to optimize the purification process using minimum steps.
Lysis was done by sonication at high amplitude in the bath sonicator for 10 min using 30 s ON, 30 s OFF cycles. Ice was used to keep the temperature low during sonication. Solubilization of inclusion bodies was optimized under various conditions. SDS-PAGE of solubilized inclusion bodies by different procedures showed that freezing inclusion bodies in the solubilizaton buffer overnight at −20 °C helped recover maximum induced protein in soluble fraction (Data not shown). Moreover, it was observed that 2M Urea is sufficient to solubilize most of the inclusion bodies (
Supplementary data Figure S1). Since some amount of residual protein was found to be present in the pellet after solubilization, varying pH, detergent supplementation, and freeze thaw cycles were tried for enhancing solubilization, but these procedures did not improve the solubilization efficiency as observed by SDS PAGE of solubilized and pellet fractions after solubilization (
Supplementary data Figure S2). Thus, an additional wash of half volume equivalent to the first wash of the solubilization buffer was given to the pellet to recover the residual induced protein. Efficacious solubilization and refolding of multiple proteins expressed in the form of inclusion bodies to obtain active proteins by this method were also reported earlier [
21]. The effect of freezing and thawing on the protein activity and stability were already described for different proteins. Slow freezing and fast thawing of proteins in solution has shown to favor maximum activity recovery and minimal damage to protein stability and activity [
22,
23]. A similar procedure was adapted for the solubilization of inclusion bodies in this study. The buffer used for solubilization employing freezing treatment also has a significant effect on the protein stability as well as buffering capacity due to the crystallization of buffer salts during freezing [
24]. Sodium phosphate shows the maximum pH change towards acidic pH upon freezing, whereas histidine, succinate, sodium acetate, and citrate buffers show an increase of less than 1 pH unit upon freezing. Tris buffer shows an increase of approximately 1.2 pH unit upon freezing [
25]. Alkaline pH has also been shown to enhance the inclusion body solubilization [
26,
27] and thus using Tris buffer proved to be an added advantage for the same since the protein is relatively stable at alkaline pH.
The solubilized protein was subjected to column purification using the
Biologic LP system and eluted in the native elution buffer using on-column protein refolding procedure.
Figure 3B shows the SDS PAGE of different fractions of the purification experiment. A single band of purified recombinant serratiopeptidase having a molecular mass of around 50 kD is seen in lane 9. The yield of the recombinant serratiopeptidase was found to be 2.5 ± 0.764 g L
−1 as confirmed by Bradford assay.
Figure 3C shows the gelatin hydrolysis activity of the elute fraction and standard serratiopeptidase. The purified recombinant serratiopeptidase showed a specific activity of 8382 ± 291 U mg
−1 protein as determined by Caseinolytic assay.
Serratiopeptidase as a therapeutic mainly finds application as an anti-inflammatory agent [
28]. Current technology for industrial-level production involves the use of a natural strain, which is evolving as an opportunistic pathogen and hence demands alternative technology for production. Reports for serratiopeptidase production are available where serratiopeptidase production has been performed using its natural strain [
29,
30,
31,
32], yielding 22.85 U/mL [
31] to 27.36 U/mL [
32] of serratiopeptidase. Although some reports of recombinant serratiopeptidase production to obtain active serratiopeptidase using
E. coli [
8,
9] and
Pichia [
33] are available, industrial production at bioreactor level requires altogether a different set of studies due to difference in the kinetics of microbial growth at the flask and bioreactor level. The serratiopeptidase yield reported in different studies using native strain and recombinant strain is described in
Table 3.
3.3. Confirmation of Recombinant Serratiopeptidase by MALDI-TOF
The identity of the pure protein was confirmed by peptide mass fingerprinting, wherein the pure protein was trypsinized and subjected to mass spectrometry. The fragmentation pattern was similar to serratiopeptidase in the first hit. Further, the identity was confirmed by MS/MS analysis, wherein the peptide sequence of one of the peaks matched with serratiopeptidase protein.
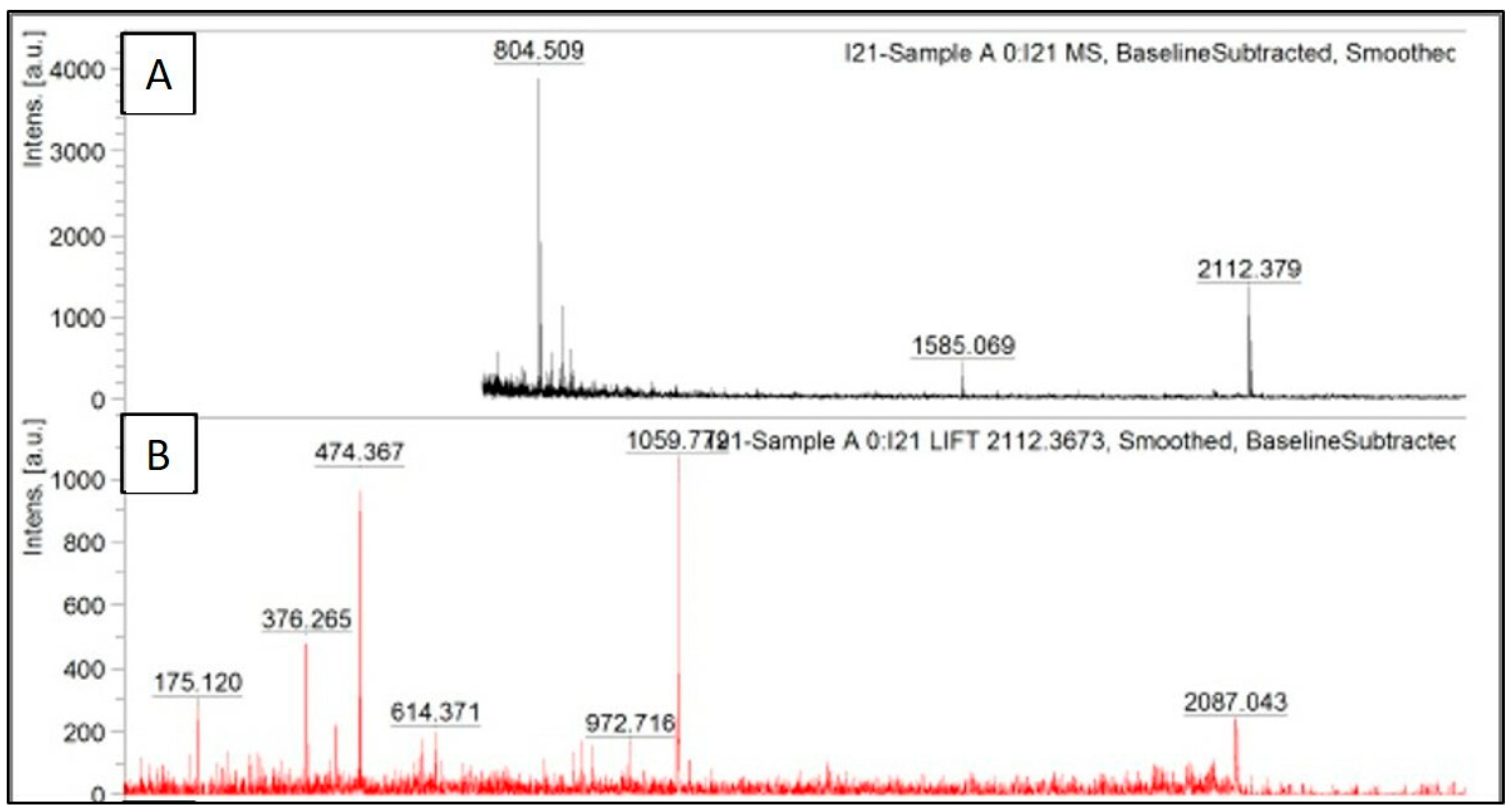
The peak data obtained after trypsinization of recombinant serratiopeptidase are shown in
Figure 4A, where the obtained masses are plotted against their relative intensities. The peak having
m/
z of 2112.3 was further subjected to MS/MS. The fragmentation pattern of the same is shown in
Figure 4B. The peak data obtained from the same was further uploaded on MASCOT database, and the protein was identified as serratiopeptidase (UNIPROT ID—P07268 (PRZN_SERME)) with a significant score of 43. The MS/MS peak data of the protein confirmed it to be serratiopeptidase with the help of MASCOT database. The MASCOT score histogram and the peak hit confirming the purified protein to be serratiopeptidase is shown in the
supplementary data (Figure S3). No peaks with
m/
z ratio similar to those obtained by
insilico trypsinization of SUMO protein were observed in MALDI TOF.
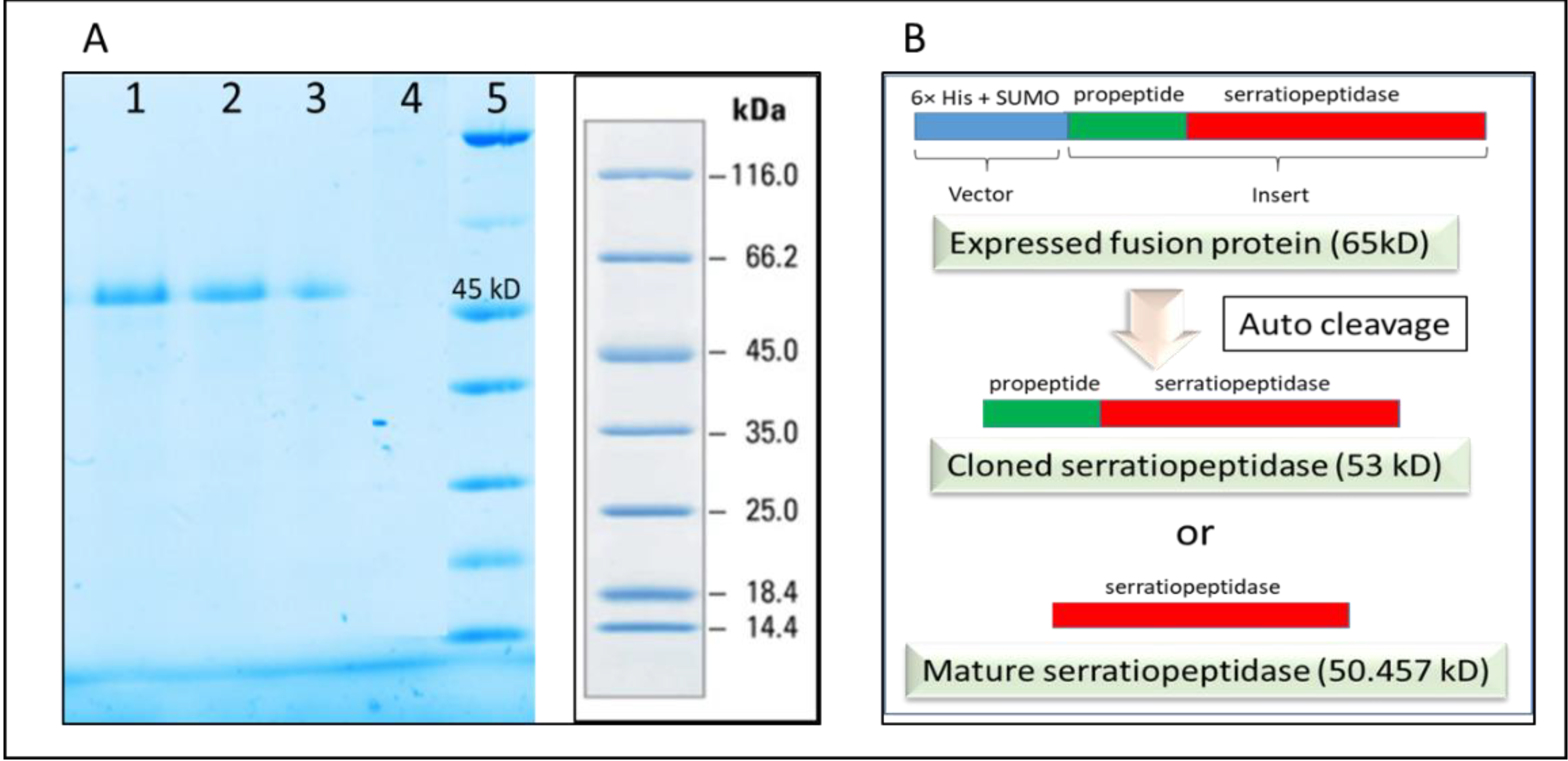
It was thus confirmed that the SUMO and thus the N terminal 6×-His tag was cleaved from purified protein after refolding. It was also confirmed by passing the purified protein through the affinity column following dialysis. The purified recombinant serratiopeptidase did not bind to the activated column and was eluted in the flow through as confirmed by SDS PAGE (
Figure 5A). Further confirmation was required for determining the exact auto cleavage site of recombinant serratiopeptidase i.e. cloned serratiopeptidase (with propeptide) or mature serratiopeptidase (without propeptide) as shown in the
Figure 5B.
3.4. Intact Mass Analysis of Purified Recombinant Serratiopeptidase by LC-MS
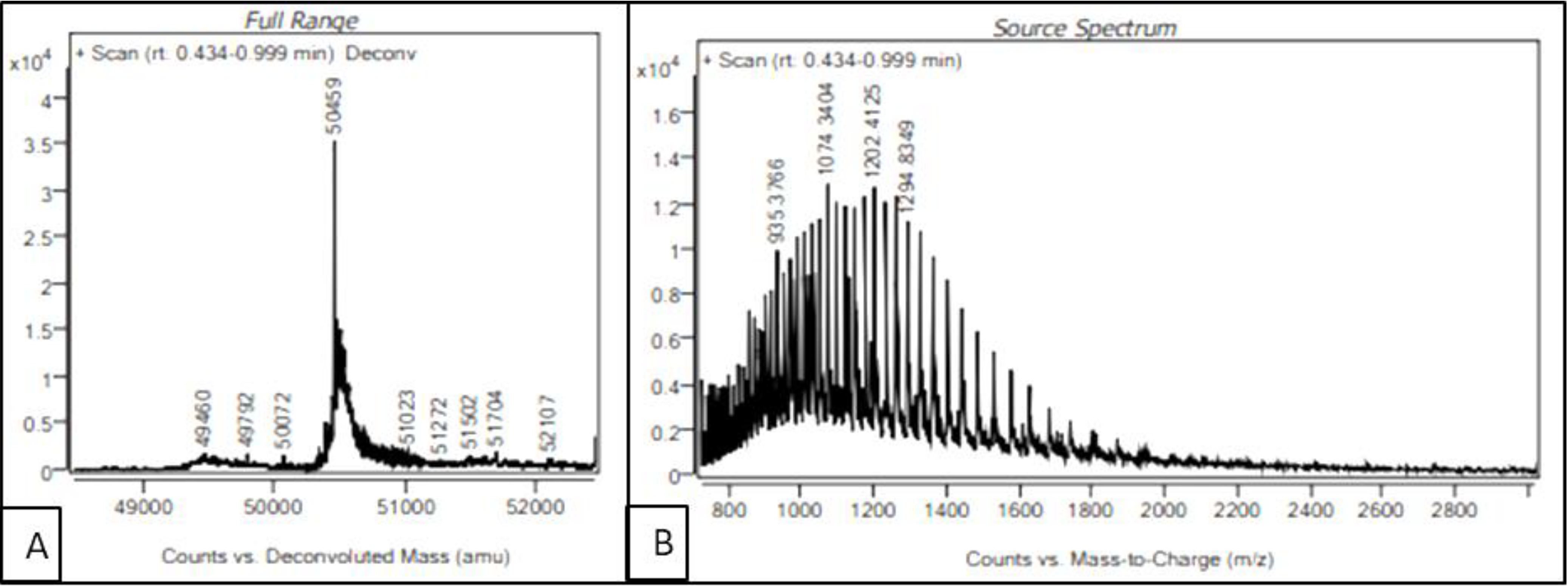
In order to identify the auto-cleavage site of the purified recombinant serratiopeptidase, intact mass of the protein was determined by LC-MS. The source spectra and deconvoluted spectra of the purified serratiopeptidase are shown in
Figure 6A,B respectively. As seen in
Figure 6A, the intact mass of the purified recombinant serratiopeptidase was estimated to be 50.459 kD, which matched the mass of mature serratiopeptidase protein without the propeptide (50.457 kD). The auto-cleavage of recombinant serratiopeptidase could be attributed to the inherent proteolytic activity of serratiopeptidase enzyme. The sequences of overexpressed recombinant serratiopeptidase and mature serratiopeptidase are given in the
supplementary data.
Serratiopeptidase belongs to a family of bacterial extracellular metalloproteases. The overexpression of serratiopeptidase was in the form of fusion protein along with the 6×-His tag, SUMO tag, and propeptide. In native strains, the propeptides are known to play a role as intramolecular chaperones that support in the correct folding of the subsequent catalytic domain and are cleaved during maturation [
36,
37]. Maturation of these metalloproteases may occur either by autocatalysis where propeptides are cleaved and degraded by the enzyme itself during maturation or by cleavage by other enzymes of the same bacteria [
38]. Such autocleavage studies have been done by [
36] on Thermolysin-like protease. Serratiopeptidase is also an extracellular metalloprotease and a similar principle of autocleavage might be leading to its cleavage at the site of propeptide upon refolding on the column. The 15 kD band might have been digested into smaller peptides by the proteolytic activity of the recombinant serratiopeptidase as the band was not seen on SDS PAGE. Also, the presence of propeptide could have a role in proper refolding of the mature serratiopeptidase because of which it is giving a higher specific activity as compared to [
8], where cloning of only mature serratiopeptidase gene was carried out. Further studies are required for confirmation.
4. Conclusions
Serratiopeptidase has shown promising results as a therapeutic agent in various studies. Threats associated with the use of the native strain have led researchers to think about alternate production strategies. As mentioned earlier, recombinant serratiopeptidase was found to be expressed in the form of inclusion bodies and could not be purified in an active form. After initial flask-level studies [
9], the cautious optimization of fed-batch procedure at the various steps of cultivation yielded pure, refolded, and active serratiopeptidase with the maximum yield per liter of media as well as maximum specific activity, to the best of our knowledge. Although the protein yield is higher than the earlier studies, there is still further scope for improvement. As mentioned earlier, the DO level, which reached below 5% during the cultivation, can be increased by aeration using air enriched with oxygen, and increasing the feed supplements with a longer batch period might still enhance the cell mass and induce a higher protein yield per batch to obtain higher cell densities as described by Korz. et al., 1995 [
16]. The self-cleavage of recombinant serratiopeptidase upon refolding, due to its proteolytic activity, is being reported for the first time, and the reason for this demands further study.



 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}