Abstract
In this study, we evaluated the modulatory effect of synbiotics (probiotics + prebiotics) on the oropharyngeal, proximal colonic, and vaginal microbiomes of Korean native pigs using 16S rRNA gene sequencing. We found increased abundances of an unclassified deltaproteobacterial genus in oropharyngeal communities of pigs supplemented with a Lactobacillus-based synbiotic. These pigs also had increased abundances of unclassified genera of Tremblayales and Lactobacillales in their proximal colons. In another group, pigs supplemented with a Bacillus-based synbiotic had increased Megasphaera and reduced Campylobacter within their oropharyngeal microbiota. In addition, their vaginal microbiota had increased Clostridium and Halalkalibacillus, as well as reduced Filifactor and Veillonella. We then explored changes in the predicted microbial functionality, associated with the synbiotics. Our analysis showed a reduction in the abundance of a fatty acid and lipid biosynthesis pathway among proximal colonic microbiomes of the Lactobacillus-fed pigs. In pigs supplemented with a Bacillus-based synbiotic, the analysis showed reduced pathway abundances for the biosynthesis of carbohydrates, as well as vitamins, cofactors, and carrier molecules within their oropharyngeal microbiomes. Meanwhile, their vaginal microbiomes had higher pathway abundances for aromatic compound degradation and secondary metabolite biosynthesis, but lower abundances for amino acid degradation. The results confirmed our hypothesis that dietary synbiotics modulate the microbiome, not only in the proximal colon, but also the oropharyngeal cavity and vaginal tract of these pigs.
1. Introduction
The concept of using probiotics to modulate microbial ecosystems as a means of promoting health and productivity in livestock has become increasingly appealing over the past two decades. This makes sense as the mammalian microbiota has been found to play important roles in the host’s nutritional, physiological, and immunological processes [1]. Moreover, the ban in many parts of the world on the use of antibiotics and zinc oxide as growth promoters has fostered the search for alternatives [2]. Excessive use and misuse of antibiotics has been implicated in the selection for, and propagation of, antimicrobial resistance (AMR) in microorganisms [3]. AMR is a global health concern, since AMR genes can be transferred between pathogens that affect, not only livestock, but also humans [4]. Besides the risk of AMR, the risk of accumulation of antibiotic residues in the food chain is another concern among consumers [5]. It is against this backdrop that probiotics, which are living microorganisms with the ability to confer health benefits to a host [6], are considered an effective and safer alternative [1].
Lactobacillus and Bacillus are some of the commonly used probiotic bacterial genera in animal feeds [1,7]. Administration of certain probiotic strains of Lactobacillus has been linked with growth promotion in pigs, partly through production of dietary enzymes such as amylases, lipases, phytases, and proteases [8]. By lowering the pH [9,10], as well as the production of bacteriocins [11], Lactobacillus strains inhibit proliferation of certain pathogens. Inclusion of Bacillus-based probiotics improves nutrient utilization in pigs, by enhancing mucosal maturity and the metabolism of lipids, carbohydrates, and proteins [12]. They also boost the pig’s immune system through stimulation of the immune system [13], production of bacteriocins and bacteriocin-like inhibitory substances [13,14], and by competitively inhibiting pathogens through promotion of lactic acid producing bacteria [15]. It is usual for probiotics to be administered together with a prebiotic (a host-indigestible dietary component, such as a fructo-oligosaccharide) a combination referred to as a synbiotic formulation [6,16]. Despite the ever-increasing interest in probiotic supplementation, their effects on the extraintestinal microbiomes remains inadequately explored. This gap is even wider in rare, understudied breeds, such as Korean native black pigs (Jeju Black Pig, JBP), which have been reared on smallholder farms on Jeju Island for generations [17].
Here, we used the 16S ribosomal RNA sequencing technique to explore the effect of in-feed synbiotics on, not only the proximal colonic, but also oropharyngeal and vaginal, microbiomes of JBP gilts. We studied two synbiotics: one consisting of three Lactobacillus strains combined with a fructo-oligosaccharide, and the second containing two Bacillus strains combined with a fructo-oligosaccharide.
2. Materials and Methods
2.1. Study Animals, Experimental Design, and Housing
This trial required 4-month-old, specific pathogen-free JBP female pigs, to evaluate the effect of Lactobacillus-based and Bacillus-based synbiotics on their microbiomes. Within these constraining specifications, the SPF facility at Cronex Co. Ltd. (Seoul, Republic of Korea) availed a total of 18 female Jeju Black Pigs (M-Pig®) from two batches of litter. Considering the 1-week age difference that existed between these 2 batches, we purposely distributed the pigs so that each treatment group had age-matched controls within the control group. In one of the treatment groups (Lactobacillus-fed, n = 4), pigs were exposed to a basal diet supplemented with a Lactobacillus-based symbiotic, while pigs in the second treatment group (Bacillus-fed, n = 5) were supplemented with a Bacillus-based synbiotic. The control group (n = 9) was allowed access to the basal diet without synbiotic supplementation.
The Lactobacillus-based synbiotic formula contained L. buchneri NLRI-1201 (1.2 × 108 colony forming units (cfu)/kg feed), L. plantarum NLRI-101 (1.6 × 108 cfu/kg feed), L. casei DK128 (1.4 × 108 cfu/kg feed), and the prebiotic, fructo-oligosaccharide (5 g/kg feed). The Bacillus-based synbiotic formula was composed of B. licheniformis DK42 (1.6 × 108 cfu/kg feed), B. subtilis SUN1234 (1.4 × 108 cfu/kg feed), and fructo-oligosaccharide (5 g/kg feed). The basal feed formulation was based on corn, soybean, and wheat.
Study animals were housed in a specific pathogen-free facility. Conditions in this facility were maintained at a temperature of 22 ± 2 °C and relative humidity of 50 ± 10%, under a 12-h light–dark cycle. The cages were designed to hold either 4 or 5 pigs, with a space allowance of 1.0 m2 per pig. Each cage was equipped with a plastic mesh floor, two round feeders, and two nipple drinkers. Throughout the experiments, the study animals were allowed ad libitum access to feed and water, and treatment commenced after a one-week acclimatization.
The gastrointestinal, urogenital, and respiratory health of the pigs was monitored daily by the caretakers and research team. Monitored parameters included appetite, diarrhea, constipation, rectal prolapses, discoloration in urine, vaginal discharge, coughing, sneezing, and rhinitis. All animals remained healthy throughout the study.
2.2. Sampling
On completion of the 12-week-long probiotic trial, the pigs were humanely euthanized, by administering pentobarbital sodium (100 mg/kg) via the external jugular vein followed by exsanguination within 30 min. Following euthanasia, swabs were aseptically collected from the oropharyngeal cavity and the vaginal canal. After dissection, samples of the luminal content in the proximal colon were collected. All samples were immediately stored at −20 °C until lab extraction of DNA was performed, within 7 days post-collection.
2.3. DNA Isolation
Total microbial community DNA was extracted from the oropharyngeal and vaginal swab samples, following the manufacturer’s instructions for the use of the QIAamp® DNA Mini kit (Qiagen, Germantown, MD, USA). To isolate total community DNA from the proximal colonic content, we used an NucleoSpin® DNA Stool Kit (Macherey-Nagel, Düren, Germany), as described in the user manual. DNA purity and concentration were evaluated using a DropSense™ 96 spectrophotometer (Trinean, Gentbrugge, Belgium).
2.4. 16S rRNA Gene Amplification and Sequencing
Hypervariable regions V3–V4 of the 16 S rRNA gene were amplified with the primers Bakt_341F and Bakt_805R [18] and used to construct NGS libraries, as previously described [19]. Amplicon libraries were pooled and sequenced on the Illumina MiSeq platform (Illumina, San Diego, CA, USA) using an MiSeq v3 reagent kit.
2.5. Bioinformatic Analysis
Sequencing yielded a total of 6,551,937 read pairs with an average of 121,332.167 read pairs per sample (range = 50,576 – 165,941). Bioinformatic analysis was performed using QIIME2 (versions, 2021.2 and 2021.11) [20]. Demultiplexed, raw fastq sequences were trimmed and quality-filtered, and paired reads were merged using DADA [21] within QIIME2 to generate amplicon sequence variants (ASVs) (Table S1). Taxonomy assignment was performed with the q2-feature-classifier plugin [22] using a Naïve Bayes classifier trained on the Greengenes reference database [23]. The classifier was taxonomically weighted to improve its accuracy on our samples, by including animal-secretion and animal-proximal-gut specific weights taken from the readytowear repository (https://github.com/BenKaehler/readytowear accessed on 14 November 2022) [24]. Chloroplast and Mitochondrial ASVs were filtered out of the dataset. After all data filtering steps, a total of 3,051,392 ASVs remained (6607 unique ASVs) with a mean of 56,507 ASVs per sample (range 12,577–81,349).
Alpha and beta diversity calculations were performed using the diversity plugin in QIIME. Prior to the diversity calculations, we chose to normalize the samples to 12,577 reads per sample, to control for uneven numbers of features per sample. Based on the alpha rarefaction curves generated, this sequencing depth was sufficient to fully observe the richness within the samples, without discarding any of the samples. For alpha diversity, we used both Faith’s phylogenetic diversity as a measure of community richness while accounting for phylogenetic relationships and Pielou’s evenness indices as a measure of community evenness. We used weighted UniFrac [25,26] and Bray–Curtis distances to compare the microbial community compositions. Statistical analysis of the observed trends in the distribution of samples among groups was tested using a permutational analysis of variance test [27] in the diversity plugin. We then predicted functional composition using PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states) implemented in the q2-picrust2 plugin [28,29].
2.6. Statistical Analysis
The output from QIIME2 was imported into R (version 4.1.3 2022-03-10), using RStudio (2022.07.0 + 353) [30] for further analysis. Statistical analysis of the alpha diversity among the study groups was performed using a Kruskal–Wallis test [31]. Pairwise comparisons between the groups were performed using Wilcoxon rank-sum tests [32], with the Benjamini–Hochberg false discovery rate (FDR) correction method used for multiple testing. Non-metric multidimensional scaling (NMDS) plots based on weighted UniFrac distance matrices were generated using the vegan [33] and ggplot2 [34] packages in R, to visualize the distribution of data among groups. Generalized linear models using a negative binomial distribution were used to test for significant associations between the taxonomic or functional composition and the synbiotic feed. We used an exact test [35] implemented in edgeR [36] to assess these associations after fitting the data to a negative binomial distribution. In all cases, statistical significance was considered at a cutoff of 0.05.
3. Results
3.1. Synbiotic Impact on Microbial Diversity
We compared the alpha diversity in the microbial communities of the three groups (Lactobacillus-based synbiotic, Bacillus-based synbiotic, and the controls). Synbiotic supplementation did not significantly influence the alpha diversity in any of the three body sites. However, the in-feed synbiotics had a significant influence on community evenness in the vaginal microbiota (Kruskal–Wallis; χ2 = 6.9667, df = 2, p-value = 0.0307). However, further analysis using a pairwise Wilcox test revealed no significant differences between the groups (Figure 1). As regards the beta diversity, the comparison of microbial communities in the three treatment groups revealed a small but significant separation of samples within the proximal colon and vaginal communities (Figure 1; PERMANOVA of Weighted-UniFrac distances in the proximal colon; pseudo-F = 1.919, p-value = 0.027; PERMANOVA of weighted-UniFrac distances in the vaginal communities; pseudo-F = 2.843, p-value = 0.001). However, the synbiotic treatment had no effect on the beta diversity within the oropharyngeal microbiota.
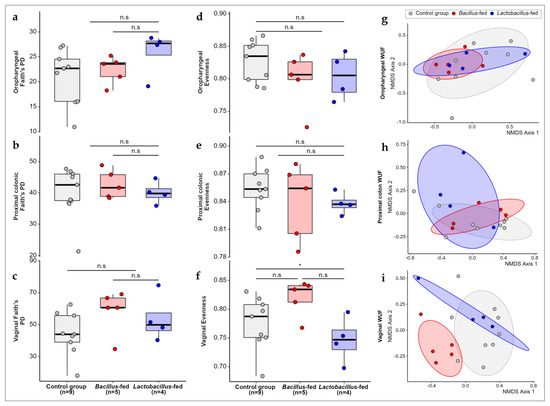
Figure 1.
Microbial diversity compared across study groups. Faith’s phylogenetic diversity among the study groups within (a) the oropharyngeal cavity, (b) proximal colon, (c) and vaginal canal. Evenness within microbial communities in the (d) oropharyngeal cavity, (e) proximal colon, and (f) and vaginal canal of the study groups. Non-metric multidimensional scaling (NMDS) plots showing weighted UniFrac distances between the microbial communities within the (g) oropharyngeal cavity, (h) proximal colon, and (i) vaginal canal of the study groups. The Asterix (*) indicates pairwise Wilcox test comparisons that were significant after correcting for multiple hypothesis testing (Benjamini–Hochberg method) at a cut-off of 0.05. Non-significant comparisons are indicated with a (n.s).
3.2. Synbiotic Impacts on Taxonomic Composition
The oropharyngeal microbiota of the pigs was dominated by Proteobacteria (35.33%), Bacteroidetes (34.57%), Fusobacteria (14.47%), and Firmicutes (10.86%) at phylum level (Table S2 and Figure S1). Proximal colonic communities were dominated by Firmicutes (65.86%) and Bacteroidetes (23.64%) (Table S3 and Figure S1), while vaginal communities predominantly consisted of Firmicutes (32.01%), Bacteroidetes (31.84%), and Proteobacteria (20.12%) (Table S4 and Figure S1). The relative abundances of phyla were not significantly altered by the synbiotics in any of the body sites. However, Bacteroidetes and Fusobacteria were differentially reduced (2-fold and 6-fold, respectively) within the vaginal microbiota of the Bacillus-fed group.
At genus level, the oropharyngeal microbiota was largely composed of Actinobacillus (9.25%), Moraxella (8.98%), Streptobacillus (8.71%), Neisseria (8.24%), Bergeyella (5.56%), and unclassified genera of the family Fusobacteriaceae (5.76%), and the order Bacteroidales (22.80%) (Table S5 and Figure S2). In the proximal colon, Streptococcus (9.68%), Lactobacillus (5.82%), and Prevotella (5.23%), as well as unclassified genera of the families Ruminococcaceae (17.62%), S24-7 (5.58%) and unclassified genera of the orders Clostridiales (17.45%) and Bacteroidales (7.40%), were predominant (Table S6 and Figure S2). Vaginal microbial communities had high relative abundances of the genera Bacteroides (5.97%), Streptococcus (5.34%), an unclassified genus of the family Pasteurellaceae (5.76%), as well as unclassified genera of the orders Bacteroidales (15.62%) and Clostridiales (7.92%) (Table S7 and Figure S2). Using an exact test, we probed for differential distribution patterns among the genera within the microbiota of the study groups. Within the oropharyngeal communities, an unclassified genus of Deltaproteobacteria was 168-fold more abundant in the microbiota of the Lactobacillus-fed group, relative to the other groups. Oropharyngeal microbial communities of the Bacillus-fed group had 68-times higher relative abundances of the genus Megasphaera and nine-times lower relative abundances of the genus Campylobacter relative to the other groups (Figure 2a and Table 1).
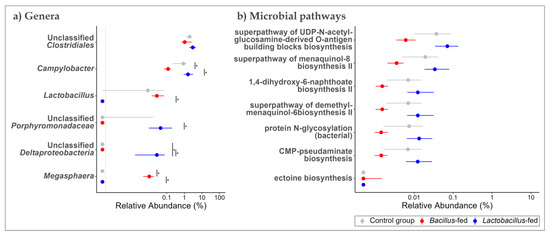
Figure 2.
Shift in the oropharyngeal microbiome across treatment groups. Genera (a) and MetaCyc microbial pathways (b) within the oropharyngeal microbiome that showed an association with at least one of the synbiotic supplements. Two methods were used here. An exact test (assuming a negative binomial distribution) to test for association with the synbiotic treatments. To test for differences in relative abundances, a Kruskal–Wallis test was used, followed by Wilcoxon rank-sum test for pairwise comparisons. The Benjamini–Hochberg (BH) FDR method was used to correct for multiple hypothesis testing, with significance considered at 0.05 (NB. plotted genera show differential distribution considering an FDR cut-off < 0.15, while the plotted microbial pathways showed differential distribution at an FDR < 0.05 and a Log FC ≥ |1|). The Asterix (*) indicates pairwise Wilcox test comparisons that were significant after correcting for multiple hypothesis testing (Benjamini–Hochberg method) at a cut-off of 0.05.

Table 1.
Differentially abundant genera in the microbiota of synbiotic-supplemented JBPs.
In the proximal colon, unclassified genera of the orders Tremblayales and Lactobacillales were differentially increased (1385-fold and 138-fold, respectively) in the Lactobacillus-fed group compared to the other groups. None of the genera were differentially shifted in the microbiota of the Bacillus-fed group (Figure 3a and Table 1).
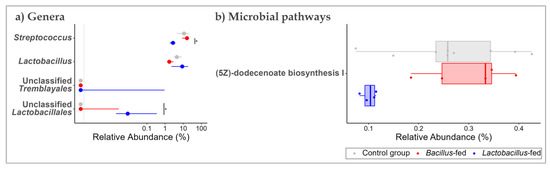
Figure 3.
Shifts in the proximal colonic microbiome across treatment groups. Genera (a) and MetaCyc microbial pathways (b) within the proximal colonic microbiome that showed an association with at least one of the synbiotic supplements. Two methods were used here: An exact test to probe for associations with each of the synbiotic treatments. To test for differences in relative abundances, a Kruskal–Wallis test was used, followed by a Wilcoxon rank-sum test for pairwise comparisons. The BH method was used to correct for multiple hypothesis testing, with significance considered at 0.05 (NB. plotted genera show differential distribution considering an FDR cut-off < 0.15, while the plotted microbial pathways show differential distribution at an FDR < 0.05 and a Log FC ≥ |1|). The Asterix (*) indicates pairwise Wilcox test comparisons that were significant after correcting for multiple hypothesis testing (Benjamini–Hochberg method) at a cut-off of 0.05.
In the vaginal microbiota, the genera Clostridium and Halalkalibacillus were differentially increased among the Bacillus-fed group (77-fold, and 139-fold, respectively). On the other hand, the Bacillus-fed group had lower relative abundances of the genera Filifactor (1363-times lower) and Veillonella (19-times) relative to other groups. In the vaginal microbiota of the Lactobacillus-fed group, the genus Haemophilus was differentially increased (1108-fold) compared to the other groups (Figure 4a and Table 1).
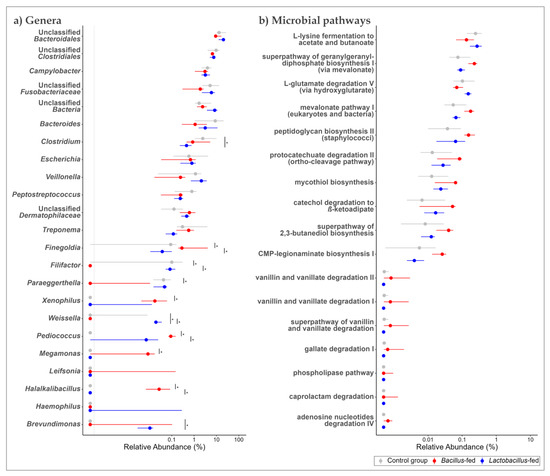
Figure 4.
Shifts in the vaginal microbiome across treatment groups. Genera (a) and MetaCyc microbial pathways (b) within the vaginal microbiome that showed an association with at least one of the synbiotic supplements. Two methods were used here: an exact test was used to explore the association between the microbial features and synbiotic treatments. To test for differences in relative abundances, a Kruskal–Wallis test was used, followed by a Wilcoxon rank-sum test for pairwise comparisons. The BH method was used to correct for multiple hypothesis testing, with significance considered at 0.05 (NB. plotted genera show a differential distribution considering an FDR cut-off < 0.15, while the plotted microbial pathways show a differential distribution at an FDR < 0.05 and a Log FC ≥ |1|). The Asterix (*) indicates pairwise Wilcox test comparisons that were significant after correcting for multiple hypothesis testing (Benjamini–Hochberg method) at a cut-off of 0.05.
3.3. Synbiotic Effect on Predicted Microbial Functionality across the Body Sites
The shifts in microbial functionality within the body sites were comparable to those detected in the microbial taxonomic composition. Again, we detected a separation of samples according to the synbiotic treatment in the microbial communities within the proximal colon and vaginal canal, but not in the oropharyngeal cavity (Figure S4, Bray–Curtis distances based on MetaCyc pathways in the proximal-colonic microbiomes, PERMANOVA pseudo-F = 2.28374, p-value = 0.035; and in vaginal microbiomes; pseudo-F = 2.660007, p-value = 0.005).
Next, we probed for microbial pathways that were significantly associated with the synbiotic treatment in the three body sites. Oropharyngeal microbiomes of the group supplemented with a Bacillus-based synbiotic were associated with an enrichment of the “ectoine biosynthesis pathway” (Figure 2b and Table 2). However, this group was also associated with a reduction of six pathways: “super pathway of UDP-N-acetylglucosamine-derived O-antigen building blocks biosynthesis”, “super-pathway of menaquinol-8 biosynthesis II”, “CMP-pseudaminate biosynthesis”, “protein N-glycosylation (bacterial)”, “1,4-dihydroxy-6-naphthoate biosynthesis II”, and the “super pathway of demethylmenaquinol-6 biosynthesis II” (Figure 2b and Table 2). In the proximal colonic microbiome, we found little effect of the synbiotic on the predicted microbial pathway abundances. Only the “(5Z)-dodecenoate biosynthesis I” pathway was reduced in the group supplemented with the Lactobacillus-based synbiotic (Figure 3b and Table 2). There were numerous pathways in the vaginal communities whose abundances showed shifts associated with synbiotic supplementation. The Bacillus-fed group had increased abundances of the pathways “caprolactam degradation”, “adenosine nucleotides degradation IV”, “phospholipase pathway”, “gallate degradation I”, “vanillin and vanillate degradation I”, “superpathway of vanillin and vanillate degradation”, “vanillin and vanillate degradation II”, “CMP-legionaminate biosynthesis I”, “catechol degradation to β-ketoadipate”, “protocatechuate degradation II (ortho-cleavage pathway)”, “peptidoglycan biosynthesis II (staphylococci)”, “superpathway of 2,3-butanediol biosynthesis”, “mycothiol biosynthesis”, “mevalonate pathway I (eukaryotes and bacteria)”, and “superpathway of geranylgeranyl diphosphate biosynthesis I (via mevalonate)” (Figure 4b and Table 2). On the other hand, the pathways “L-lysine fermentation to acetate and butanoate” and “L-glutamate degradation V (via hydroxyglutarate)” were associated with a reduction in abundance within the vaginal microbiomes of the group supplemented with a Bacillus-based synbiotic (Figure 4b and Table 2).
Table 2.
Differentially abundant pathways in the microbiome of symbiotic-supplemented JBPs.
4. Discussion
This study assessed the impact of Lactobacillus-based and Bacillus-based synbiotic supplements on the oropharyngeal, proximal colonic, and vaginal microbiomes of Korean native black pigs.
Our results indicate that the synbiotics had no effect on the alpha diversity within these communities but explained the clustering observed between samples from the proximal colonic and vaginal communities. We also uncovered some compositional and functional features of these communities that were significantly associated with the synbiotic treatment. Overall, the Lactobacillus-based synbiotics had more pronounced effects on the proximal colonic microbiome, while the Bacillus-based synbiotics showed stronger effects on the vaginal communities.
Although the synbiotics did not alter the alpha and beta diversity in oropharyngeal microbial communities, they influenced the relative abundances of some taxa. For example, an unclassified genus of the class Deltaproteobacteria was associated with an increased relative abundance in the Lactobacillus-fed group. Members of this class are largely characterized by their ability to reduce sulfates and sulfites [37], producing hydrogen sulfide (H2S), which has widespread and varied effects on the host’s physiology [38,39]. This metabolite influences several processes, including inflammation, apoptosis, and nociception, among others, although the reported effects seem contradictory under the various conditions explored in the literature [39,40]. Among the group fed a Bacillus-based synbiotic, we found an association with increased abundances of Megasphaera and reduced abundances of Campylobacter. Megasphaera plays a role in the immune and inflammatory response of the host through production of short chain fatty acid (SCFA) metabolites, such as butyrate. Butyrate is known to modulate the immune response through various mechanisms, including influencing immune cell recruitment, chemokine production, and dendritic cell activation [41,42]. Butyrate is mostly important in the lower GIT but also has effects in other body sites, such as the respiratory tract. For example, butyrate’s anti-inflammatory effects have been demonstrated to alleviate acute bacterial pneumonias [43,44]. The genus Campylobacter includes several pathobionts of the oral cavity, and gastrointestinal and reproductive tracts [45]. An inhibitory effect on potential pathogens from this genus would be beneficial to the pigs. There is a glaring lack of studies exploring the impact of probiotics on the oropharyngeal microbiota. Some studies have explored the role of probiotics, including those containing Lactobacillus spp., on oropharyngeal and/or upper respiratory tract health [46,47]. In these studies, however, it was hypothesized that the detected effects were triggered by probiotic effects within the GIT [48,49,50]. Nonetheless, our data provide evidence of a modulatory effect in the oropharyngeal microbiomes of these pigs following synbiotic supplementation in their diets.
Interestingly, we found only two taxa whose relative abundances were significantly associated with the synbiotics in the proximal colon. These taxa included unclassified genera of the orders Tremblayales and Lactobacillales, which were associated with increased relative abundances within the proximal colonic microbial communities of the Lactobacillus-fed group. Sequences assigned to the Tremblayales order belong to enigmatic taxa that phylogenetically align with members of the class Betaproteobacteria based on 16S rRNA gene sequence data. These organisms have been detected in environmental samples but are closely related to the better-known tiny obligate intracellular symbiotic bacteria, Candidatus Tremblaya spp., which intriguingly contain even smaller endosymbionts of the Gammaproteobacteria class [51,52,53]. In the porcine gut, the Tremblayales group has been found to occur in increasingly high abundances among pigs with high body weight [54]. Members of the order Lactobacillales are mainly defined by their ability to yield lactic acid as the main by-product of carbohydrate fermentation [55]. Lactobacillales play important roles in the host’s defense against gastrointestinal pathogens. This can be through a lactic-acid-dependent disruption of bacterial cell walls [10], lowering of pH [56], or production of bacteriocins [11,57]. Lactobacillales also exhibit an immunomodulatory activity through stimulation of cytokine expression [58,59]. The increase in the members of the order Lactobacillales within the group supplemented with the Lactobacillus-based synbiotic was of no surprise. This is because supplementation with any Lactobacillus spp.-based probiotics [9,60] and fructo-oligosaccharide prebiotics [61] has been found to stimulate the proliferation of members of the Lactobacillales.
We found several genera within the vaginal microbiome of the JBP to be significantly associated with dietary supplementation using the Bacillus-based synbiotics. In particular, the genera Clostridium and Halalkalibacillus were increased within this group, while Filifactor and Veillonella were reduced. Clostridium spp. are a butyrogenic group of microbes [62]. Although butyrate is known to be important in the GIT, its role in the reproductive tract remains relatively understudied. There is some evidence, however, that butyrate may have a regulatory role in the secretion of estrogen and progesterone by the ovarian granulosa cells [63]. Through their β-glucuronidase enzymatic activity, Clostridium spp. also produce biologically active catecholamines, including noradrenaline and dopamine [64]. These are neurotransmitters of the sympathetic nervous system that predominantly control uterine contractility and consequently embryo migration and implantation [65,66,67]. Halalkalibacillus are halophilic and alkaliphilic, spore-forming bacteria that were first isolated from non-saline soils [68]. While there have been no reports on this genus within the vaginal microbiota of pigs, other members of the family Bacillaceae have been reported to have higher abundances among gilts compared to sows [69]. The reduction in the relative abundances of Filifactor that we detected might imply the potential for Bacillus-based probiotics to enhance the reproductive performance of pigs, since this genus has been linked to low reproductive performance in sows [70]. The genus is also linked to negative impacts in other livestock species such as cows, where it has been found to be a predictor of metritis within the uterine microbiota of cows [71]. The other significantly reduced genus, Veillonella, metabolizes lactate [72,73]. Its reduction in the vaginal microbiota of the Bacillus-fed group was surprising, since the lactate-yielding genus Lactobacillus was significantly increased in these communities (Figure S2). Abundance of vaginal Veillonella spp. is, however, associated with a reduced risk for perineal organ prolapse in sows during gestation [74]. Only one genus within the vaginal communities, Haemophilus, was significantly associated with the Lactobacillus-based synbiotic. Proliferation of Haemophilus spp. is generally considered to require hemin (factor X) and/or nicotinamide adenine dinucleotide (NAD) (factor V) [75]. Haemophilus spp. are well known pathobionts of the upper respiratory tract in several mammalian species, including pigs [75]. Their presence in the vaginal microbiota is not well documented in swine, but several species of this genus have been isolated from the vaginal tracts of humans [76] and cows [77]. Overall, several taxa varied significantly in these communities. A plausible explanation for this is that the high diversity and low evenness within this community makes it more prone to extensive compositional shifts following perturbation.
In addition to the compositional changes, we identified predicted metabolic pathways across the body sites that were associated with synbiotic supplementation. Within the oropharyngeal microbiomes, the predicted pathways involved in the biosynthesis of carbohydrates, as well as vitamins, cofactors, and carrier molecules, were reduced among the Bacillus-fed group. In particular, we found a reduction in the potential for synthesis of carbohydrates, such as lipopolysaccharides [78,79], which might suggest a reduction in Gram-negative bacteria, some of which are pathogenic. Another group of pathways that were reduced included those involved in the biosynthesis of ubiquinone and menaquinone, which are essential components of the electron transport chain in aerobic bacteria [80]. Reduction of these pathways, therefore, might point to a relative reduction in the abundances of aerobic bacteria within the oropharyngeal microbiota of the Bacillus-fed pigs. Furthermore, there was an increased potential for biosynthesis of ectoine, which is an osmoregulatory cyclic amino acid that protects bacteria against stressors in the form of osmotic pressure, temperature, and free radicals, among others [81]. This suggests a link between Bacillus-supplementation and the proliferation of stress tolerant microbial taxa within oropharyngeal microbiomes.
While the beta diversity among proximal colonic microbiota showed differences across treatments, we only observed a few taxa that were significantly associated with the Lactobacillus-based synbiotic. Consistently with the few changes in phylogenetic composition, at functional level, only the “(5Z)-dodecenoate biosynthesis I pathway” was significantly reduced in these communities. This pathway is involved in fatty acid oxidation, yielding a medium chain fatty acid metabolite, (5Z)-dodecenoate. This metabolite has been linked to inhibition of fungal proliferation, by inhibiting hyphae growth [82]. A reduced potential for (5Z)-dodecenoate biosynthesis, associated with the Lactobacillus-based synbiotic, is therefore likely to disrupt the colonic mycobiome–bacteriome balance, leading to opportunistic GIT yeast infections.
Supplementation with the Bacillus-based synbiotic was associated with shifts in the abundances of several pathways in the vaginal microbiome. Among these was an increase in several pathways involved in aromatic compound degradation. Degradation of aromatic compounds by microorganisms is a well-studied phenomenon due to its bioremediation potential, as these compounds are common environmental pollutants, due to their wide application in manufacturing [83]. Degradation pathways for caprolactam, vanillin, and protocatechuate, as with other aromatic compounds, are enriched in wastewater and rhizosphere microbiomes, and this has been hypothesized to be due to the presence of xenobiotic pollutants in soils [84]. The reason for the enrichment of these pathways in the vaginal microbiome of our JBP gilts is unclear. However, a recent study reported similar findings within the vaginal microbiome of cattle [85]. The authors attributed the proliferation of these pathways to a possible colonization by the microbiome originating from the floors within the housing environment. While our animals were housed in a controlled environment, this cannot be ruled out as a possible explanation. Another possible explanation is that Bacterial species with the potential for aromatic compound degradation within the fecal microbiota of the Bacillus-fed group might have made their way from the perineal area into the vaginal canal. Indeed, Bacillus spp. such as those used in the synbiotic are known to possess the potential for aromatic compound degradation [86]. Another relatively increased pathway among the Bacillus-fed group was involved in the biosynthesis of mycothiol, which is a protective thiol in the ecologically ubiquitous Actinobacteria [87]. Mycothiol not only protects the producing organism against oxidative stress, but also degrades xenobiotics [87]. The increased abundance of this pathway was probably due to the increased abundance of the Actinobacteria among the pigs supplemented with a Bacillus-based synbiotic (Figure S1). We also detected an increased abundance of two pathways involved in secondary metabolite biosynthesis. These pathways are involved in the biosynthesis of geranylgeranyl diphosphate, which is a C20 linear precursor for a variety of diterpenoids [88]. Bacterial secondary metabolites such as diterpenes and other terpenoids are thought to have ecological roles for the organism or strain that produces them [89]. In this ecological context, they are used for signaling and competition during inter- and intra-specific microbial interactions within their environment. Meanwhile, within the context of host health, their bacteriostatic and bactericidal activity could contribute towards the inhibition of potential pathogens of the porcine urogenital tract. Indeed, the properties of terpenes are diverse and are being intensively explored for pharmaceutical applications [90]. On the other hand, the Bacillus-fed group was associated with a relatively low abundances of predicted pathways involved in amino acid degradation, suggesting a reduction in the prevalence of obligate amino acid fermenting anaerobes [91,92]. Indeed, we did detect a relative reduction in abundances of obligate amino acid fermenting taxa such as Clostridium, Peptostreptococcus, and Fusobacteria within the vaginal microbiota of the Bacillus-fed pigs (Table 1).
Probiotics have had a growing application in the maintenance of sexual and reproductive health in women [93]. Although their application in the clinical management of gynecological and obstetric conditions is feasible and a preferable alternative to antimicrobials, its efficacy remains inconclusive so far [94]. As such, further research remains necessary and the use of the pig as a biomedical animal model facilitates such extensive studies, given that pigs have physiological and anatomical similarities to humans [95]. Our study explored an oral route of probiotic application, which assumes that some of the ingested probiotic organisms can transit through the gastrointestinal tract and ascend into the vaginal tract from the rectum. Although this is more plausible in the pig than it is in humans, for behavioral reasons, changes in the vaginal microbiome have been noted following oral probiotic regimens in women [96]. Another mechanism through which oral probiotics could influence vaginal microbiota is through indirect systemic effects, due to the probiotic’s modulatory effects within the gut microbiome [97,98]. Although it has been intuitively suggested that a topical, intra-vaginal application route may yield clearer results, both routes of administration have been found to be effective [99]. Overall, orally administered probiotics warrant attention, since they are convenient to use and likely, more agreeable. Such a non-invasive application route, provides opportunities for the development of vaginal-microbiome-restoring probiotics following gynecological procedures, especially when fertility sparing is a goal [93,100].
5. Conclusions
Our study indicated that Bacillus- and Lactobacillus-based synbiotic supplementation was associated with shifts in the taxonomic composition and functionality within the three microbial communities explored in the JBPs. Although the alpha diversity in these body sites was not significantly shifted, analysis of the beta diversities indicated that the clustering in proximal colonic and vaginal samples was significantly explained by the synbiotic supplementation. Additionally, the synbiotics were associated with shifts in the abundances of several microbial taxa and the predicted pathways within these body sites.
These findings suggest that synbiotic supplementation may have microbiome-mediated effects, not only in the GIT (proximal colon), but also in the oropharyngeal cavity and vaginal canal of pigs. However, this study had a few limitations, including the following: First, the inclusion criteria for the experimental animals greatly limited the group sizes used in this study, which slightly underpowered our findings. Second, our work was based on a metataxonomic approach, and the shifts in predicted microbial functionality described herein only represent changes in potential functionality. To validate our findings, future studies with larger sample sizes should combine the metataxonomic strategy used here with meta-transcriptomic and metaproteomic approaches, which can identify the differential expression of microbial pathways and proteins.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/fermentation9040359/s1, Figure S1: Dominant phyla in the oropharyngeal proximal colonic and vaginal microbiome; Figure S2: Dominant genera in oropharyngeal, proximal colonic, and vaginal microbiome; Table S1: Denoising statistics of the sequences that passed the quality filtering process; Table S2: Dominant phyla in oropharyngeal microbiota of the JBP gilts; Table S3: Dominant phyla in the proximal colonic microbiota of the JBP gilts; Table S4: Dominant phyla in the vaginal microbiota of the JBP gilts; Table S5: Dominant genera in the oropharyngeal microbiota of the JBP gilts; Table S6: Dominant genera in the proximal colonic microbiota of the JBP gilts; Table S7: Dominant genera in the vaginal microbiota of the JBP gilts.
Author Contributions
Conceptualization, A.W.B., K.-D.S., H.-K.L. and J.H.; methodology, A.W.B., K.-D.S., H.-K.L. and J.H.; software, A.W.B.; validation: A.W.B., K.-D.S. and J.H.; formal analysis: A.W.B. and J.H.; investigation: A.W.B., K.-D.S. and J.H.; resources: K.-D.S. and J.H.; data curation: A.W.B. and J.H.; writing—original draft preparation: A.W.B.; writing—review & editing: A.W.B., K.-D.S., H.-K.L. and J.H.; visualization: A.W.B.; supervision: J.H.; project administration: K.-D.S. and J.H.; funding acquisition: J.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by grants from the Next-Generation BioGreen 21 Program (PJ01322302), Rural Development Administration, Republic of Korea, and the National Research Foundation of Korea (NRF) funded by the Korea government (MSIT) (No. 2021K1A3A9A05019142).
Institutional Review Board Statement
All animal handling procedures in this study followed the ARRIVE guidelines (https://arriveguidelines.org) and were approved by Cronex Co., Ltd.’s Animal Care and Use Committee (CRONEX-IACUC 202002007).
Informed Consent Statement
Not applicable.
Data Availability Statement
Raw sequences are available through the Sequence Read Archive (SRA) with accession number PRJNA926012 (https://www.ncbi.nlm.nih.gov/bioproject/926012).
Acknowledgments
We thank all staff at the Cronex Animal SPF facility for their support in animal care, data, and sample collection.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kenny, M.; Smidt, H.; Mengheri, E.; Miller, B. Probiotics–do they have a role in the pig industry? Animal 2011, 5, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Maron, D.F.; Smith, T.J.; Nachman, K.E. Restrictions on antimicrobial use in food animal production: An international regulatory and economic survey. Glob. Health 2013, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- WHO. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Lusk, J.L.; Norwood, F.B.; Pruitt, J.R. Consumer demand for a ban on antibiotic drug use in pork production. Am. J. Agric. Econ. 2006, 88, 1015–1033. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef]
- Markowiak, P.; Śliżewska, K. The role of probiotics, prebiotics and synbiotics in animal nutrition. Gut Pathog. 2018, 10, 21. [Google Scholar] [CrossRef]
- Kim, E.-Y.; Kim, Y.-H.; Rhee, M.-H.; Song, J.-C.; Lee, K.-W.; Kim, K.-S.; Lee, S.-P.; Lee, I.-S.; Park, S.-C. Selection of Lactobacillus sp. PSC101 that produces active dietary enzymes such as amylase, lipase, phytase and protease in pigs. J. Gen. Appl. Microbiol. 2007, 53, 111–117. [Google Scholar] [CrossRef]
- Konstantinov, S.R.; Smidt, H.; Akkermans, A.D.; Casini, L.; Trevisi, P.; Mazzoni, M.; De Filippi, S.; Bosi, P.; De Vos, W.M. Feeding of Lactobacillus sobrius reduces Escherichia coli F4 levels in the gut and promotes growth of infected piglets. FEMS Microbiol. Ecol. 2008, 66, 599–607. [Google Scholar] [CrossRef]
- Alakomi, H.-L.; Skytta, E.; Saarela, M.; Mattila-Sandholm, T.; Latva-Kala, K.; Helander, I. Lactic acid permeabilizes gram-negative bacteria by disrupting the outer membrane. Appl. Environ. Microbiol. 2000, 66, 2001–2005. [Google Scholar] [CrossRef]
- Cotter, P.D.; Hill, C.; Ross, R.P. Bacteriocins: Developing innate immunity for food. Nat. Rev. Microbiol. 2005, 3, 777–788. [Google Scholar] [CrossRef]
- He, Y.; Jinno, C.; Kim, K.; Wu, Z.; Tan, B.; Li, X.; Whelan, R.; Liu, Y. Dietary Bacillus spp. enhanced growth and disease resistance of weaned pigs by modulating intestinal microbiota and systemic immunity. J. Anim. Sci. Biotechnol. 2020, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.A.; Duc, L.H.; Cutting, S.M. The use of bacterial spore formers as probiotics. FEMS Microbiol. Rev. 2005, 29, 813–835. [Google Scholar] [CrossRef] [PubMed]
- Urdaci, M.; Pinchuk, I. Antimicrobial activity of Bacillus probiotics. In Bacterial Spore Formers–Probiotics Emerging Applications; Horizon Bioscience: Norfolk, UK, 2004; pp. 171–182. [Google Scholar]
- Hosoi, T.; Ametani, A.; Kiuchi, K.; Kaminogawa, S. Improved growth and viability of lactobacilli in the presence of Bacillus subtilis (natto), catalase, or subtilisin. Can. J. Microbiol. 2000, 46, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Olveira, G.; González-Molero, I. An update on probiotics, prebiotics and symbiotics in clinical nutrition. Endocrinol. Y Nutr. 2016, 63, 482–494. [Google Scholar] [CrossRef]
- Cho, I.-C.; Han, S.-H.; Fang, M.; Lee, S.-S.; Ko, M.-S.; Lee, H.; Lim, H.-T.; Yoo, C.-K.; Lee, J.-H.; Jeon, J.-T. The robust phylogeny of Korean wild boar (Sus scrofa coreanus) using partial D-loop sequence of mtDNA. Mol. Cells 2009, 28, 423–430. [Google Scholar] [CrossRef]
- Herlemann, D.P.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef]
- Bugenyi, A.W.; Lee, M.-R.; Choi, Y.-J.; Song, K.-D.; Lee, H.-K.; Son, Y.-O.; Lee, D.-S.; Lee, S.-C.; Son, Y.-J.; Heo, J. Oropharyngeal, proximal colonic, and vaginal microbiomes of healthy Korean native black pig gilts. BMC Microbiol. 2023, 23, 3. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Kaehler, B.D.; Bokulich, N.A.; McDonald, D.; Knight, R.; Caporaso, J.G.; Huttley, G.A. Species abundance information improves sequence taxonomy classification accuracy. Nat. Commun. 2019, 10, 4643. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Wilcoxon, F. Individual Comparisons by Ranking Methods. In Breakthroughs in Statistics; Springer: Berlin/Heidelberg, Germany, 1992; pp. 196–202. [Google Scholar]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Wickham, H. Data Analysis. In ggplot2; Springer: Berlin/Heidelberg, Germany, 2016; pp. 189–201. [Google Scholar]
- Robinson, M.D.; Smyth, G.K. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 2008, 9, 321–332. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.L.; Kjeldsen, K.U.; Rattei, T.; Pester, M.; Loy, A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi) sulfite reductases. ISME J. 2015, 9, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Sen, N. Functional and molecular insights of hydrogen sulfide signaling and protein sulfhydration. J. Mol. Biol. 2017, 429, 543–561. [Google Scholar] [CrossRef]
- Li, L.; Moore, P. Putative biological roles of hydrogen sulfide in health and disease: A breath of not so fresh air? Trends Pharmacol. Sci. 2008, 29, 84–90. [Google Scholar] [CrossRef]
- Singh, S.B.; Lin, H.C. Hydrogen sulfide in physiology and diseases of the digestive tract. Microorganisms 2015, 3, 866–889. [Google Scholar] [CrossRef] [PubMed]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Vieira, R.d.S.; Castoldi, A.; Basso, P.J.; Hiyane, M.I.; Câmara, N.O.S.; Almeida, R.R. Butyrate attenuates lung inflammation by negatively modulating Th9 cells. Front. Immunol. 2019, 10, 67. [Google Scholar] [CrossRef]
- Ni, Y.-F.; Wang, J.; Yan, X.-L.; Tian, F.; Zhao, J.-B.; Wang, Y.-J.; Jiang, T. Histone deacetylase inhibitor, butyrate, attenuates lipopolysaccharide-induced acute lung injury in mice. Respir. Res. 2010, 11, 33. [Google Scholar] [CrossRef]
- Man, S.M. The clinical importance of emerging Campylobacter species. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 669–685. [Google Scholar] [CrossRef]
- De Vrese, M.; Winkler, P.; Rautenberg, P.; Harder, T.; Noah, C.; Laue, C.; Ott, S.; Hampe, J.; Schreiber, S.; Heller, K. Effect of Lactobacillus gasseri PA 16/8, Bifidobacterium longum SP 07/3, B. bifidum MF 20/5 on common cold episodes: A double blind, randomized, controlled trial. Clin. Nutr. 2005, 24, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Berggren, A.; Lazou Ahrén, I.; Larsson, N.; Önning, G. Randomised, double-blind and placebo-controlled study using new probiotic lactobacilli for strengthening the body immune defence against viral infections. Eur. J. Nutr. 2011, 50, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Yitbarek, A.; Taha-Abdelaziz, K.; Hodgins, D.C.; Read, L.; Nagy, É.; Weese, J.S.; Caswell, J.L.; Parkinson, J.; Sharif, S. Gut microbiota-mediated protection against influenza virus subtype H9N2 in chickens is associated with modulation of the innate responses. Sci. Rep. 2018, 8, 13189. [Google Scholar] [CrossRef]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; Peaper, D.R.; Ho, J.H.; Murray, T.S.; Iwasaki, A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5354–5359. [Google Scholar] [CrossRef]
- Dang, A.T.; Marsland, B.J. Microbes, metabolites, and the gut–lung axis. Mucosal Immunol. 2019, 12, 843–850. [Google Scholar] [CrossRef]
- Von Dohlen, C.D.; Kohler, S.; Alsop, S.T.; McManus, W.R. Mealybug β-proteobacterial endosymbionts contain γ-proteobacterial symbionts. Nature 2001, 412, 433–436. [Google Scholar] [CrossRef]
- Thao, M.L.; Gullan, P.J.; Baumann, P. Secondary (γ-Proteobacteria) endosymbionts infect the primary (β-Proteobacteria) endosymbionts of mealybugs multiple times and coevolve with their hosts. Appl. Environ. Microbiol. 2002, 68, 3190–3197. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.P.; Moran, N.A. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 2012, 10, 13–26. [Google Scholar] [CrossRef]
- Oh, J.K.; Chae, J.P.; Pajarillo, E.A.B.; Kim, S.H.; Kwak, M.J.; Eun, J.S.; Chee, S.W.; Whang, K.Y.; Kim, S.H.; Kang, D.K. Association between the body weight of growing pigs and the functional capacity of their gut microbiota. Anim. Sci. J. 2020, 91, e13418. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E.V. Evolutionary genomics of lactic acid bacteria. J. Bacteriol. 2007, 189, 1199–1208. [Google Scholar] [CrossRef]
- Fayol-Messaoudi, D.; Berger, C.N.; Coconnier-Polter, M.-H.; Lievin-Le Moal, V.; Servin, A.L. pH-, Lactic acid-, and non-lactic acid-dependent activities of probiotic Lactobacilli against Salmonella enterica Serovar Typhimurium. Appl. Environ. Microbiol. 2005, 71, 6008–6013. [Google Scholar] [CrossRef] [PubMed]
- Twomey, D.; Ross, R.; Ryan, M.; Meaney, B.; Hill, C. Lantibiotics produced by lactic acid bacteria: Structure, function and applications. Antonie Van Leeuwenhoek 2002, 82, 165–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Yu, H.; Gao, X.; Li, X.; Qiao, S. Influence of Lactobacillus fermentum I5007 on the intestinal and systemic immune responses of healthy and E. coli challenged piglets. Antonie Van Leeuwenhoek 2009, 96, 89–98. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; O’Regan, P.; Fanning, Á.; O’Mahony, C.; MacSharry, J.; Lyons, A.; Bienenstock, J.; O’Mahony, L.; Shanahan, F. Functional modulation of human intestinal epithelial cell responses by Bifidobacterium infantis and Lactobacillus salivarius. Immunology 2006, 118, 202–215. [Google Scholar] [CrossRef]
- Ohashi, Y.; Tokunaga, M.; Taketomo, N.; Ushida, K. Stimulation of indigenous lactobacilli by fermented milk prepared with probiotic bacterium, Lactobacillus delbrueckii subsp. bulgaricus strain 2038, in the pigs. J. Nutr. Sci. Vitaminol. 2007, 53, 82–86. [Google Scholar] [CrossRef]
- Konstantinov, S.R.; Awati, A.; Smidt, H.; Williams, B.A.; Akkermans, A.D.; De Vos, W.M. Specific response of a novel and abundant Lactobacillus amylovorus-like phylotype to dietary prebiotics in the guts of weaning piglets. Appl. Environ. Microbiol. 2004, 70, 3821–3830. [Google Scholar] [CrossRef]
- Willems, A.; Amat-Marco, M.; Collins, M.D. Phylogenetic analysis of Butyrivibrio strains reveals three distinct groups of species within the Clostridium subphylum of the gram-positive bacteria. Int. J. Syst. 1996, 46, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Li, M.; Lei, H.; Jiang, X.; Tu, W.; Lu, Y.; Xia, D. Butyric acid regulates progesterone and estradiol secretion via cAMP signaling pathway in porcine granulosa cells. J. Steroid Biochem. Mol. Biol. 2017, 172, 89–97. [Google Scholar] [CrossRef]
- Asano, Y.; Hiramoto, T.; Nishino, R.; Aiba, Y.; Kimura, T.; Yoshihara, K.; Koga, Y.; Sudo, N. Critical role of gut microbiota in the production of biologically active, free catecholamines in the gut lumen of mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2012, 303, G1288–G1295. [Google Scholar] [CrossRef]
- Taneike, T.; Narita, T.; Kitazawa, T.; Bando, S.; Teraoka, H.; Ohga, A. Binding and functional characterization of alpha-2 adrenoceptors in isolated swine myometrium. J. Auton. Pharmacol. 1995, 15, 93–105. [Google Scholar] [CrossRef]
- Kitazawa, T.; Nakagoshi, K.; Teraoka, H.; Taneike, T. 5-HT7 receptor and β2-adrenoceptor share in the inhibition of porcine uterine contractility in a muscle layer-dependent manner. Eur. J. Pharmacol. 2001, 433, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Fanchin, R.; Righini, C.; Olivennes, F.; Taylor, S.; de Ziegler, D.; Frydman, R. Uterine contractions at the time of embryo transfer alter pregnancy rates after in-vitro fertilization. Hum. Reprod. 1998, 13, 1968–1974. [Google Scholar] [CrossRef] [PubMed]
- Echigo, A.; Fukushima, T.; Mizuki, T.; Kamekura, M.; Usami, R. Halalkalibacillus halophilus gen. nov., sp. nov., a novel moderately halophilic and alkaliphilic bacterium isolated from a non-saline soil sample in Japan. Int. J. Syst. Evol. Microbiol. 2007, 57, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Luque, A.T.; Fontana, C.; Pasteris, S.E.; Bassi, D.; Cocconcelli, P.S.; Otero, M.C. Vaginal bacterial diversity from healthy gilts and pregnant sows subjected to natural mating or artificial insemination. Res. Vet. Sci. 2021, 140, 26–37. [Google Scholar] [CrossRef]
- Sanglard, L.; Schmitz-Esser, S.; Gray, K.; Linhares, D.; Yeoman, C.; Dekkers, J.; Niederwerder, M.; Serão, N. Vaginal microbiota diverges in sows with low and high reproductive performance after porcine reproductive and respiratory syndrome vaccination. Sci. Rep. 2020, 10, 3046. [Google Scholar] [CrossRef]
- Jeon, S.J.; Vieira-Neto, A.; Gobikrushanth, M.; Daetz, R.; Mingoti, R.D.; Parize, A.C.B.; de Freitas, S.L.; da Costa, A.N.L.; Bicalho, R.C.; Lima, S. Uterine microbiota progression from calving until establishment of metritis in dairy cows. Appl. Environ. Microbiol. 2015, 81, 6324–6332. [Google Scholar] [CrossRef]
- Rogosa, M. THE GENUS VEILLONELLA I: General Cultural, Ecological, and Biochemical Considerations. Carbohydr. Res. 1964, 87, 162–170. [Google Scholar]
- Rogosa, M.; Bishop, F.S. THE GENUS VEILLONELLA II: Nutritional Studies. J. Bacteriol. 1964, 87, 574–580. [Google Scholar] [CrossRef]
- Kiefer, Z.E.; Koester, L.R.; Studer, J.M.; Chipman, A.L.; Mainquist-Whigham, C.; Keating, A.F.; Schmitz-Esser, S.; Ross, J.W. Vaginal microbiota differences associated with pelvic organ prolapse risk during late gestation in commercial sows. Biol. Reprod. 2021, 105, 1545–1561. [Google Scholar] [CrossRef]
- Fink, D.L.; Geme, J.W. The Genus Haemophilus. In The Prokaryotes: A Handbook on the Biology of Bacteria Volume 6: Proteobacteria: Gamma Subclass; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 1034–1061. [Google Scholar]
- Messing, M.; Sottnek, F.O.; Biddle, J.W.; Schlater, L.K.; Kramer, M.A.; Kraus, S.J. Isolation of Haemophilus species from the genital tract. Sex. Transm. Dis. 1983, 10, 56–61. [Google Scholar] [CrossRef]
- Kwiecien, J.M.; Little, P.B. Haemophilus somnus and reproductive disease in the cow: A review. Can. Vet. J. 1991, 32, 595. [Google Scholar] [PubMed]
- Schoenhofen, I.C.; McNally, D.J.; Brisson, J.-R.; Logan, S.M. Elucidation of the CMP-pseudaminic acid pathway in Helicobacter pylori: Synthesis from UDP-N-acetylglucosamine by a single enzymatic reaction. Glycobiology 2006, 16, 8C–14C. [Google Scholar] [CrossRef] [PubMed]
- Samuel, G.; Reeves, P. Biosynthesis of O-antigens: Genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr. Res. 2003, 338, 2503–2519. [Google Scholar] [CrossRef]
- Bentley, R.; Meganathan, R. Biosynthesis of vitamin K (menaquinone) in bacteria. Microbiol. Rev. 1982, 46, 241–280. [Google Scholar] [CrossRef]
- Pastor, J.M.; Salvador, M.; Argandoña, M.; Bernal, V.; Reina-Bueno, M.; Csonka, L.N.; Iborra, J.L.; Vargas, C.; Nieto, J.J.; Cánovas, M. Ectoines in cell stress protection: Uses and biotechnological production. Biotechnol. Adv. 2010, 28, 782–801. [Google Scholar] [CrossRef] [PubMed]
- Seelbinder, B.; Chen, J.; Brunke, S.; Vazquez-Uribe, R.; Santhaman, R.; Meyer, A.-C.; de Oliveira Lino, F.S.; Chan, K.-F.; Loos, D.; Imamovic, L. Antibiotics create a shift from mutualism to competition in human gut communities with a longer-lasting impact on fungi than bacteria. Microbiome 2020, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.-S.; Keum, Y.-S.; Li, Q.X. Bacterial degradation of aromatic compounds. Int. J. Environ. Res. Public Health 2009, 6, 278–309. [Google Scholar] [CrossRef]
- Yurgel, S.N.; Nearing, J.T.; Douglas, G.M.; Langille, M.G. Metagenomic functional shifts to plant induced environmental changes. Front. Microbiol. 2019, 10, 1682. [Google Scholar] [CrossRef]
- Ong, C.T.; Ross, E.M.; Boe-Hansen, G.; Turni, C.; Hayes, B.J.; Fordyce, G.; Tabor, A.E. Adaptive sampling during sequencing reveals the origins of the bovine reproductive tract microbiome across reproductive stages and sexes. Sci. Rep. 2022, 12, 15075. [Google Scholar] [CrossRef]
- Kazunga, C.; Aitken, M.D. Products from the incomplete metabolism of pyrene by polycyclic aromatic hydrocarbon-degrading bacteria. Appl. Environ. Microbiol. 2000, 66, 1917–1922. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.L.; Buchmeier, N.; Fahey, R.C. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. [Google Scholar] [CrossRef]
- Smanski, M.J.; Peterson, R.M.; Huang, S.-X.; Shen, B. Bacterial diterpene synthases: New opportunities for mechanistic enzymology and engineered biosynthesis. Curr. Opin. Chem. Biol. 2012, 16, 132–141. [Google Scholar] [CrossRef]
- Tyc, O.; Song, C.; Dickschat, J.S.; Vos, M.; Garbeva, P. The ecological role of volatile and soluble secondary metabolites produced by soil bacteria. Trends Microbiol. 2017, 25, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Ajikumar, P.K.; Tyo, K.; Carlsen, S.; Mucha, O.; Phon, T.H.; Stephanopoulos, G. Terpenoids: Opportunities for biosynthesis of natural product drugs using engineered microorganisms. Mol. Pharm. 2008, 5, 167–190. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.O.; Russell, J.B. An rRNA approach for assessing the role of obligate amino acid-fermenting bacteria in ruminal amino acid deamination. Appl. Environ. Microbiol. 1996, 62, 815–821. [Google Scholar] [CrossRef]
- Buckel, W.; Barker, H.A. Two pathways of glutamate fermentation by anaerobic bacteria. J. Bacteriol. 1974, 117, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Lehtoranta, L.; Ala-Jaakkola, R.; Laitila, A.; Maukonen, J. Healthy vaginal microbiota and influence of probiotics across the female life span. Front. Microbiol. 2022, 13, 787. [Google Scholar] [CrossRef]
- Buggio, L.; Somigliana, E.; Borghi, A.; Vercellini, P. Probiotics and vaginal microecology: Fact or fancy? BMC Women’s Health 2019, 19, 25. [Google Scholar] [CrossRef]
- Pang, X.; Hua, X.; Yang, Q.; Ding, D.; Che, C.; Cui, L.; Jia, W.; Bucheli, P.; Zhao, L. Inter-species transplantation of gut microbiota from human to pigs. ISME J. 2007, 1, 156–162. [Google Scholar] [CrossRef]
- Chen, C.; Hao, L.; Zhang, Z.; Tian, L.; Zhang, X.; Zhu, J.; Jie, Z.; Tong, X.; Xiao, L.; Zhang, T. Cervicovaginal microbiome dynamics after taking oral probiotics. J. Genet. Genom. 2021, 48, 716–726. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Chai, J.N.; Tianero, M.D.; Di Luccia, B.; Ahern, P.P.; Merriman, J.; Cortez, V.S.; Caparon, M.G.; Donia, M.S.; Gilfillan, S. Lactobacillus reuteri induces gut intraepithelial CD4+ CD8αα+ T cells. Science 2017, 357, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, T.; Li, Y.; Zhang, T.; Wang, Q.; He, J.; Wang, L.; Li, L.; Yang, N.; Fang, Y. Probiotics for the treatment of women with bacterial vaginosis: A systematic review and meta-analysis of randomized clinical trials. Eur. J. Pharmacol. 2019, 864, 172660. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, A.; Luisa, F.M.; Dellino, M.; Erica, S.; Loizzi, V.; Bocciolone, L.; Rabaiotti, E.; Cioffi, R.; Sabetta, G.; Cormio, G. Fertility sparing surgery in sex-cord stromal tumors: Oncological and reproductive outcomes. Int. J. Gynecol. Cancer 2022, 32, 1063–1070. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).