
Extracellular Production of the Taiwan-Native Norovirus P Domain Overexpressed in Pichia pastoris
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Media
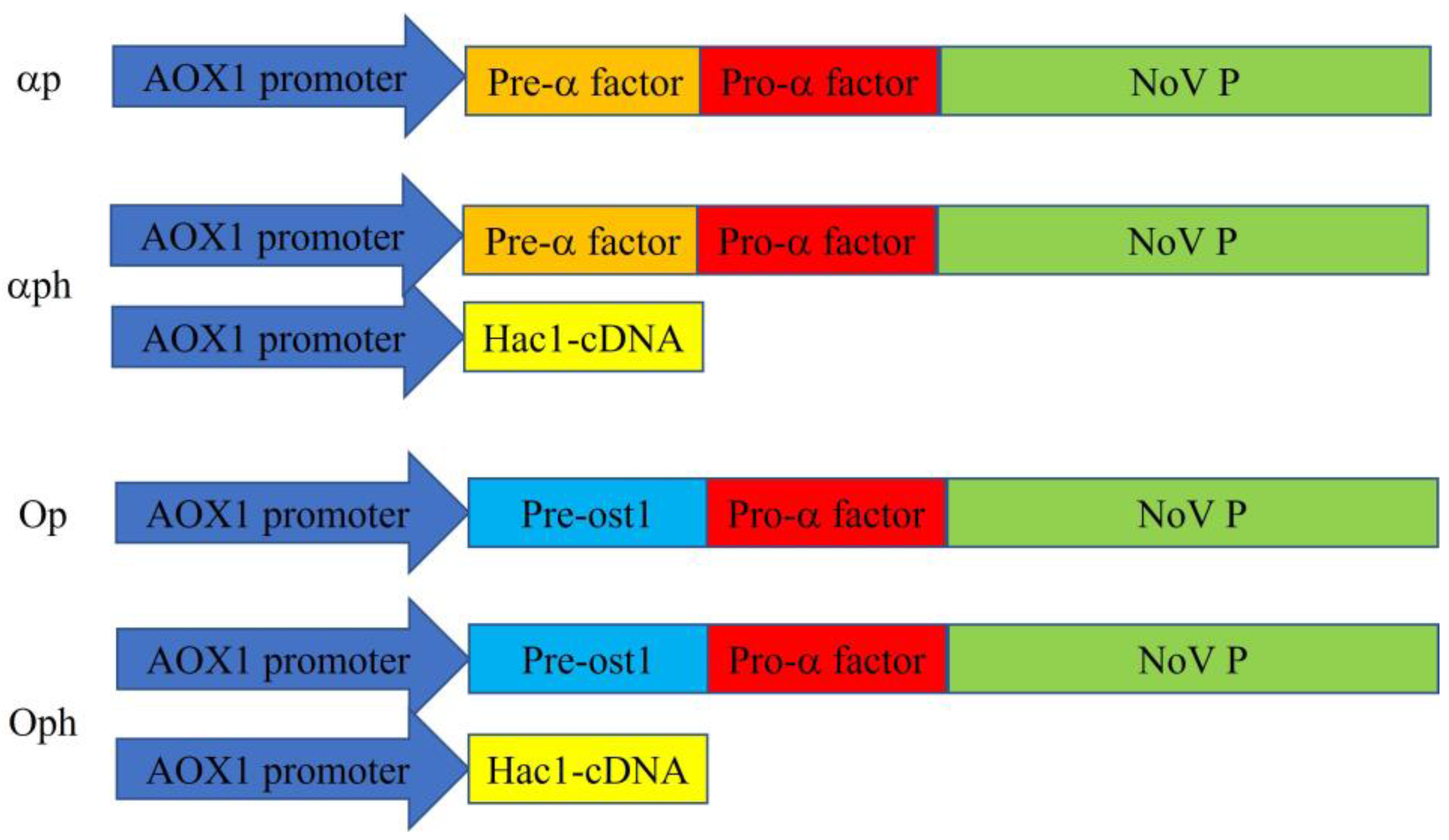
2.2. Cloning of the P Domain and Plasmid Construction
2.3. Transformant Screening and Flask Culture
2.4. AOX Activity Assay
2.5. RNA Expression Level Analysis
2.6. Fermentation
2.7. Purification of NoV P Protein
2.8. Gel Filtration Analysis
2.9. SDS–PAGE and Western Blot Analysis
2.10. Indirect ELISA
2.11. Saliva-Binding Assay
2.12. Identification of NoV P by LC-MS/MS
2.13. Particle Size Analysis and Transmission Electron Microscopy (TEM)
2.14. Statistical Analysis
3. Results
3.1. Expression of NoV P Protein from Flask Cultures
3.2. NoV P Protein Production in Fermenter Cultures
3.3. NoV P Protein Verification by LC-MS/MS
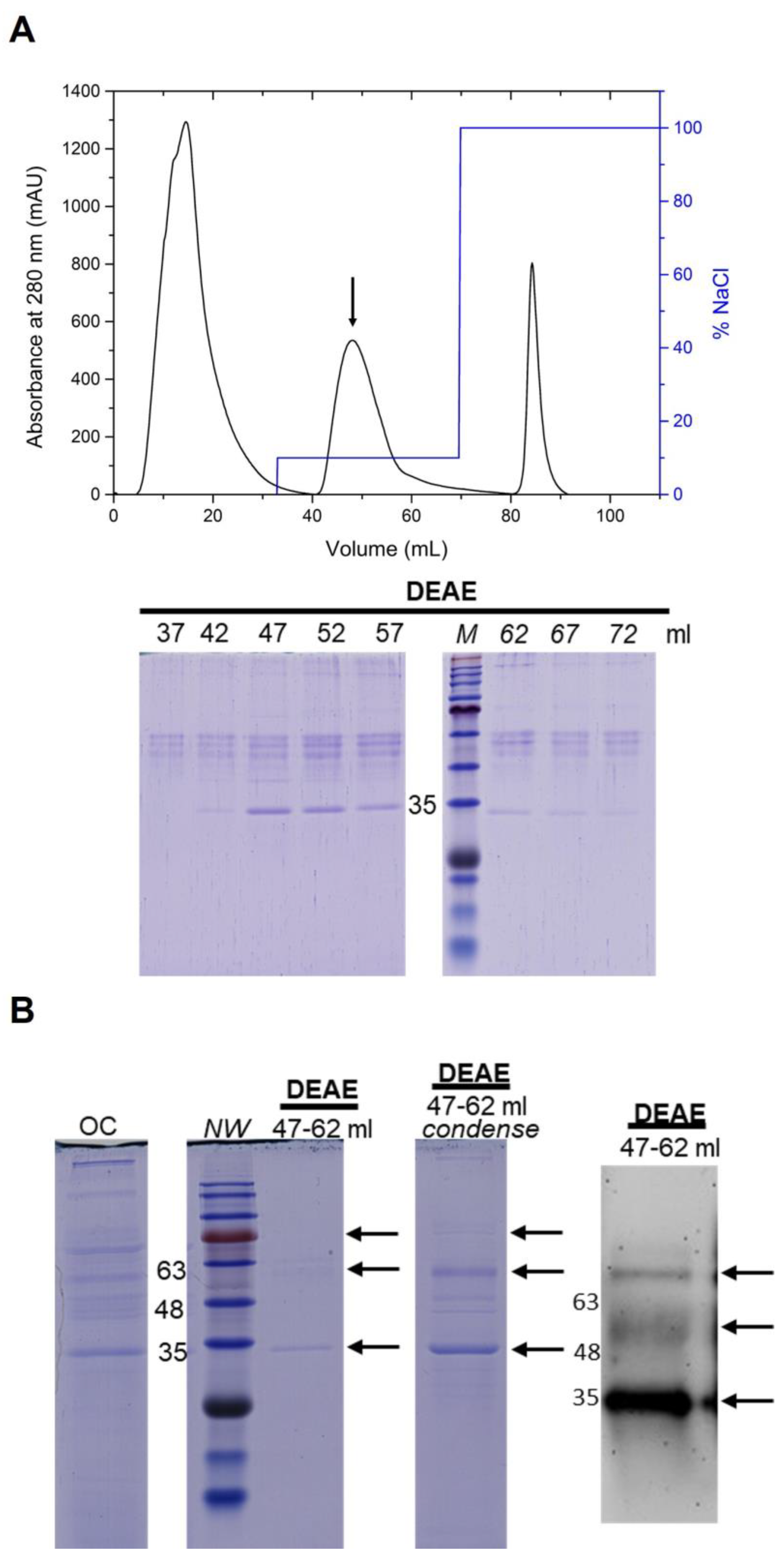
3.4. Purification of the NoV P Protein by Anion-Exchange Chromatography
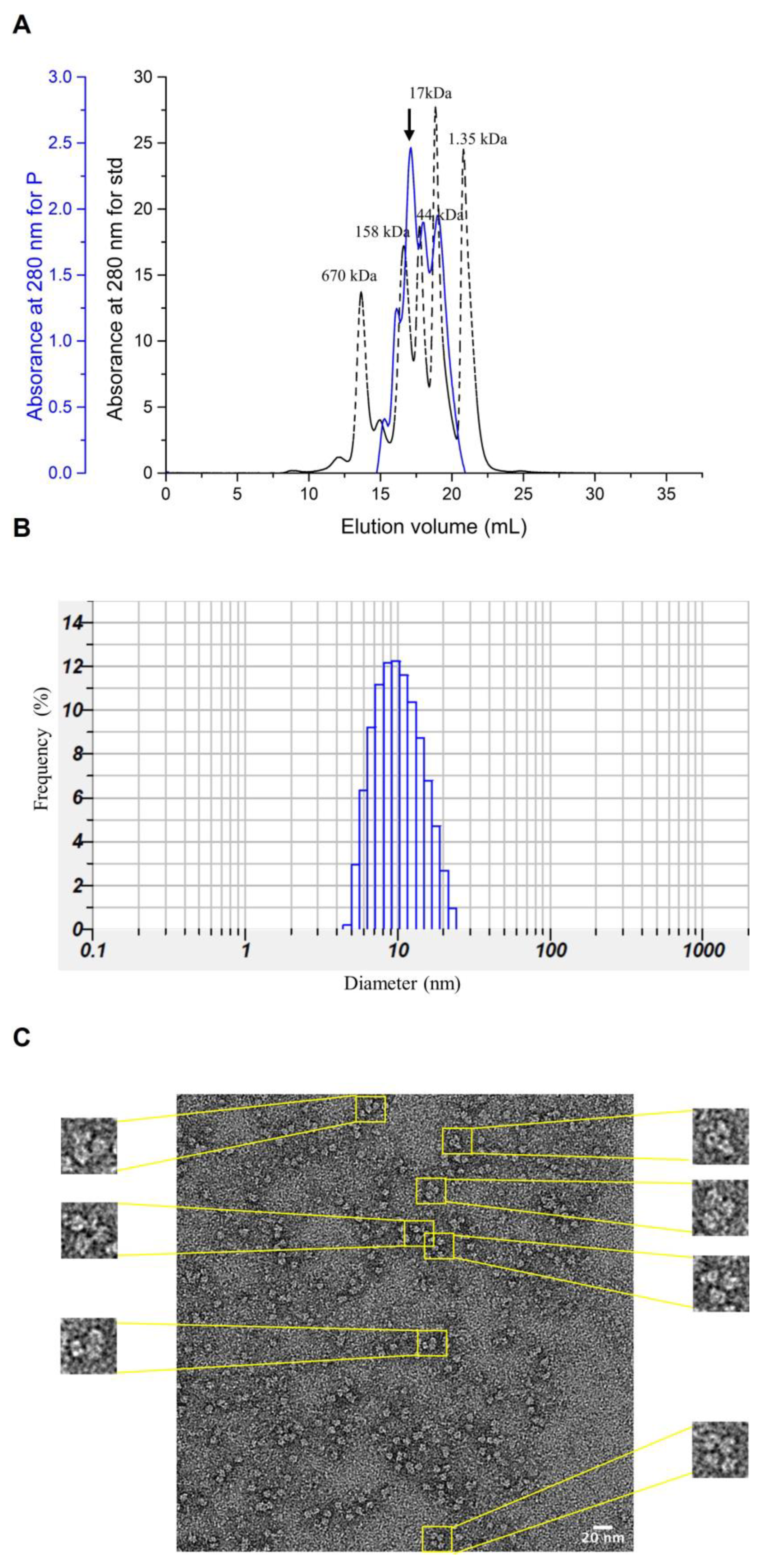
3.5. Characterization of Purified NoV P Complexes and Verification by TEM
3.6. Saliva-Binding Assay of NoV P
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, M.M.; Widdowson, M.-A.; Glass, R.I.; Akazawa, K.; Vinje, J.; Parashar, U.D. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 2008, 14, 1224–1231. [Google Scholar] [CrossRef]
- Li, J.; Predmore, A.; Divers, E.; Lou, F. New interventions against human norovirus: Progress, opportunities, and challenges. Annu. Rev. Food Sci. Technol. 2012, 3, 331–352. [Google Scholar] [CrossRef]
- Lucero, Y.; Vidal, R.; O’Ryan, G.M. Norovirus vaccines under development. Vaccine 2018, 36, 5435–5441. [Google Scholar] [CrossRef]
- Takanashi, S.; Saif, L.J.; Hughes, J.H.; Meulia, T.; Jung, K.; Scheuer, K.A.; Wang, Q. Failure of propagation of human norovirus in intestinal epithelial cells with microvilli grown in three-dimensional cultures. Arch. Virol. 2014, 159, 257–266. [Google Scholar] [CrossRef]
- Parra, G.I.; Bok, K.; Taylor, R.; Haynes, J.R.; Sosnovtsev, S.V.; Richardson, C.; Green, K.Y. Immunogenicity and specificity of norovirus Consensus GII.4 virus-like particles in monovalent and bivalent vaccine formulations. Vaccine 2012, 30, 3580–3586. [Google Scholar] [CrossRef]
- El-Kamary, S.S.; Pasetti, M.F.; Mendelman, P.M.; Frey, S.E.; Bernstein, D.I.; Treanor, J.J.; Ferreira, J.; Chen, W.H.; Sublett, R.; Richardson, C.; et al. Adjuvanted intranasal Norwalk virus-like particle vaccine elicits antibodies and antibody-secreting cells that express homing receptors for mucosal and peripheral lymphoid tissues. J. Infect. Dis. 2010, 202, 1649–1658. [Google Scholar] [CrossRef]
- Tan, M.; Huang, P.; Xia, M.; Fang, P.-A.; Zhong, W.; McNeal, M.; Wei, C.; Jiang, W.; Jiang, X. Norovirus P particle, a novel platform for vaccine development and antibody production. J. Virol. 2011, 85, 753–764. [Google Scholar] [CrossRef]
- Fang, H.; Tan, M.; Xia, M.; Wang, L.; Jiang, X. Norovirus P particle efficiently elicits innate, humoral and cellular immunity. PLoS ONE 2013, 8, e63269. [Google Scholar] [CrossRef]
- Xia, M.; Wei, C.; Wang, L.; Cao, D.; Meng, X.-J.; Xiang, X.; Tan, M. A trivalent vaccine candidate against hepatitis E virus, norovirus, and astrovirus. Vaccine 2016, 34, 905–913. [Google Scholar] [CrossRef]
- Xia, M.; Tan, M.; Wei, C.; Zhong, W.; Wang, L.; McNeal, M.; Jiang, X. A candidate dual vaccine against influenza and noroviruses. Vaccine 2011, 29, 7670–7677. [Google Scholar] [CrossRef]
- Tan, M.; Jiang, X. The p domain of norovirus capsid protein forms a subviral particle that binds to histo-blood group antigen receptors. J. Virol. 2005, 79, 14017–14030. [Google Scholar] [CrossRef]
- Tamminen, K.; Huhti, L.; Koho, T.; Lappalainen, S.; Hytönen, V.P.; Vesikari, T.; Blazevic, V. A comparison of immunogenicity of norovirus GII-4 virus-like particles and P-particles. Immunology 2012, 135, 89–99. [Google Scholar] [CrossRef]
- Tomé-Amat, J.; Fleisher, L.; Parker, S.A.; Bardliving, C.; Batt, C.A. Secreted production of assembled Norovirus virus-like particles from Pichia pastoris. Microb. Cell Factories 2014, 13, 134. [Google Scholar] [CrossRef]
- Tan, M.; Jiang, X. Norovirus P particle: A subviral nanoparticle for vaccine development against norovirus, rotavirus and influenza virus. Nanomedicine 2012, 7, 889–897. [Google Scholar] [CrossRef]
- Fu, L.; Jin, H.; Yu, Y.; Yu, B.; Zhang, H.; Wu, J.; Yin, Y.; Yu, X.; Wu, H.; Kong, W. Characterization of NoV P particle-based chimeric protein vaccines developed from two different expression systems. Protein Expr. Purif. 2017, 130, 28–34. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chang, P.J.; Huang, C.T. Small P particles formed by the Taiwan-native norovirus P domain overexpressed in Komagataella pastoris. Appl. Microbiol. Biotechnol. 2018, 102, 9707–9718. [Google Scholar] [CrossRef]
- Chen, Y.L.; Huang, C.T. Establishment of a two-step purification scheme for tag-free recombinant Taiwan native norovirus P and VP1 proteins. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1159, 122357. [Google Scholar] [CrossRef]
- Ahmad, M.; Hirz, M.; Pichler, H.; Schwab, H. Protein expression in Pichia pastoris: Recent achievements and perspectives for heterologous protein production. Appl. Microbiol. Biotechnol. 2014, 98, 5301–5317. [Google Scholar] [CrossRef]
- Brake, A.J.; Merryweather, J.P.; Coit, D.G.; Heberlein, U.A.; Masiarz, F.R.; Mullenbach, G.T.; Urdea, M.S.; Valenzuela, P.; Barr, P.J. Alpha-factor-directed synthesis and secretion of mature foreign proteins in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1984, 81, 4642–4646. [Google Scholar] [CrossRef]
- Delic, M.; Valli, M.; Graf, A.B.; Pfeffer, M.; Mattanovich, D.; Gasser, B. The secretory pathway: Exploring yeast diversity. FEMS Microbiol. Rev. 2013, 37, 872–914. [Google Scholar] [CrossRef]
- Idiris, A.; Tohda, H.; Kumagai, H.; Takegawa, K. Engineering of protein secretion in yeast: Strategies and impact on protein production. Appl. Microbiol. Biotechnol. 2010, 86, 403–417. [Google Scholar] [CrossRef]
- Fitzgerald, I.; Glick, B.S. Secretion of a foreign protein from budding yeasts is enhanced by cotranslational translocation and by suppression of vacuolar targeting. Microb. Cell Fact. 2014, 13, 125. [Google Scholar] [CrossRef]
- Barrero, J.J.; Casler, J.C.; Valero, F.; Ferrer, P.; Glick, B.S. An improved secretion signal enhances the secretion of model proteins from Pichia pastoris. Microb. Cell Fact. 2018, 17, 161. [Google Scholar] [CrossRef]
- Duan, G.; Ding, L.; Wei, D.; Zhou, H.; Chu, J.; Zhang, S.; Qian, J. Screening endogenous signal peptides and protein folding factors to promote the secretory expression of heterologous proteins in Pichia pastoris. J. Biotechnol. 2019, 306, 193–202. [Google Scholar] [CrossRef]
- Guerfal, M.; Ryckaert, S.; Jacobs, P.P.; Ameloot, P.; Van Craenenbroeck, K.; Derycke, R.; Callaewert, N. The HAC1 gene from Pichia pastoris: Characterization and effect of its overexpression on the production of secreted, surface displayed and membrane proteins. Microb. Cell Fact. 2010, 9, 49. [Google Scholar] [CrossRef]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Zhao, X.; Xie, W.; Lin, Y.; Lin, X.; Zheng, S.; Han, S. Combined strategies for improving the heterologous expression of an alkaline lipase from Acinetobacter radioresistens CMC-1 in Pichia pastoris. Process Biochem. 2013, 48, 1317–1323. [Google Scholar] [CrossRef]
- Stasyk, O.V.; Nazarko, T.Y.; Sibirny, A.A. Methods of plate pexophagy monitoring and positive selection for ATG gene cloning in yeasts. Methods Enzymol. 2008, 451, 229–239. [Google Scholar]
- Chang, C.-H.; Hsiung, H.-A.; Hong, K.-L.; Huang, C.-T. Enhancing the efficiency of the Pichia pastoris AOX1 promoter via the synthetic positive feedback circuit of transcription factor Mxr1. BMC Biotechnol. 2018, 18, 81. [Google Scholar] [CrossRef]
- Barrero, J.J.; Pagazartaundua, A.; Glick, B.S.; Valero, F.; Ferrer, P. Bioreactor-scale cell performance and protein production can be substantially increased by using a secretion signal that drives co-translational translocation in Pichia pastoris. N. Biotechnol. 2021, 60, 85–95. [Google Scholar] [CrossRef]
- Rieder, L.; Ebner, K.; Glieder, A.; Sørlie, M. Novel molecular biological tools for the efficient expression of fungal lytic polysaccharide monooxygenases in Pichia pastoris. Biotechnol. Biofuels 2021, 14, 122. [Google Scholar] [CrossRef]
- Li, L.; Huang, C.; Zhao, F.; Deng, T.; Lin, Y.; Zheng, S.; Liang, S.; Han, S. Improved production and characterization of Volvariella volvacea Endoglucanase 1 expressed in Pichia pastoris. Protein Expr. Purif. 2018, 152, 107–113. [Google Scholar] [CrossRef]
- Tan, M.; Jiang, X. Norovirus Capsid Protein-Derived Nanoparticles and Polymers as Versatile Platforms for Antigen Presentation and Vaccine Development. Pharmaceutics 2019, 11, 472. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′ to 3′) |
|---|---|
| F α-factor | CGTGTCTTCTGCTGCTCCAGTCAACACTACAACAG |
| R EcoRI α-factor | GAATTCAGCTTCAGCC |
| F BstBI Ost1 | TTCGAAACGATGAGGCAGGTTTGGTTCTCTTGGATTGTGGGATTG |
| R Ost1 | TCTCTTGGATTGTGGGATTGTTCCTATGTTTTTTCAACGTGTCTTCTGCTGCTCCAG |
| F EcoRI CNGRC P | GAATTCATGTGTAATGGTCGTTGTTCAAGAACTAAAC |
| R XbaI ORF2 stop | TCTAGATTATAAAGCACGTCTACGCC |
| F EcoRI HAC1 | GAATTCATGCCCGTAGATTCTTCTCATAAGA |
| R XhoI HAC1 | CTCGAGCTATTCCTGGAAGAATACAAAGTCATTTAAAT |
| Name | Sequence (5′ to 3′) |
|---|---|
| 18s rRNA qF | GAGGATTGACAGGATGAGAGC |
| 18s rRNA qR | CAAGGTCTCGTTCGTTATCGC |
| HAC1 qF | GACACCGACTACATTACTACAGCTCCA |
| HAC1 qR | AGCGGTAAATGGTGCTGCTGG |
| Adsorbent | P Protein (μg/mL) | Total Protein (μg/mL) | Purity (%) | P Recovery (%) |
|---|---|---|---|---|
| Supernatant | 345.2 ± 35.1 | 520.6 ± 25.6 | 66.1 ± 3.4 | |
| DEAE Sepharose FF Stepwise elution 10% | 228.9 ± 3.0 | 241.8 ± 13.7 | 94.8 ± 4.1 | 66.9 ± 5.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chien, M.-L.; Yu, C.-F.; Huang, C.-T. Extracellular Production of the Taiwan-Native Norovirus P Domain Overexpressed in Pichia pastoris. Fermentation 2023, 9, 498. https://doi.org/10.3390/fermentation9060498
Chien M-L, Yu C-F, Huang C-T. Extracellular Production of the Taiwan-Native Norovirus P Domain Overexpressed in Pichia pastoris. Fermentation. 2023; 9(6):498. https://doi.org/10.3390/fermentation9060498
Chicago/Turabian StyleChien, Man-Ling, Chun-Fu Yu, and Ching-Tsan Huang. 2023. "Extracellular Production of the Taiwan-Native Norovirus P Domain Overexpressed in Pichia pastoris" Fermentation 9, no. 6: 498. https://doi.org/10.3390/fermentation9060498