
Integrated Metabolome and Transcriptome Analyses Reveal That the Flavonoid Metabolic Pathway Is Associated with Pigment Differential Accumulation in Two Colors of Petaloid Staminodes in Canna glauca
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
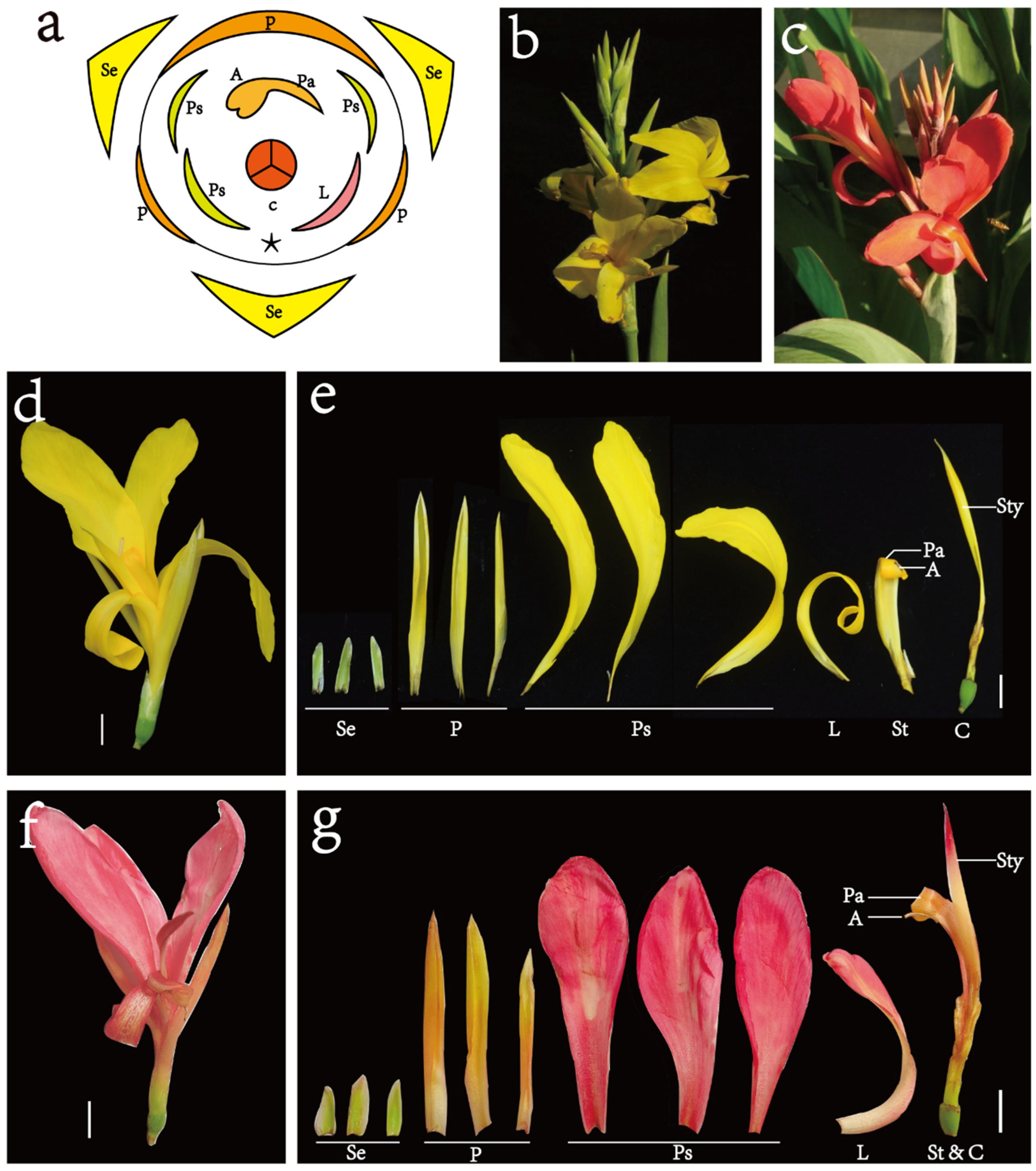
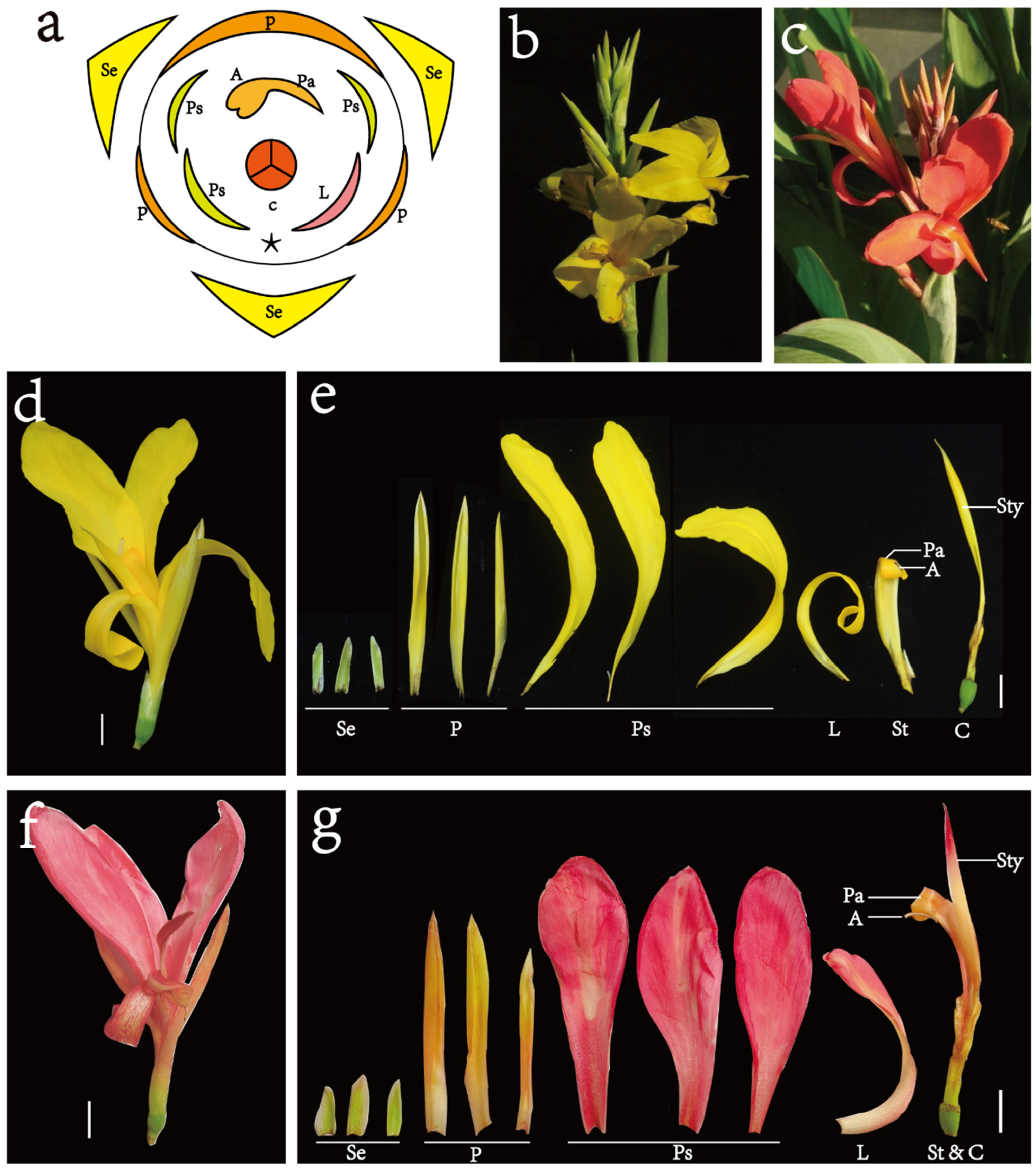
2.1. Plants
2.2. Flavonoid Metabolite Extraction and Analysis
2.3. Differential Metabolite Identification
2.4. Transcriptome Sequencing
2.5. Gene Annotation and Differentially Expressed Gene Identification
2.6. Real-Time Quantitative PCR
2.7. Correlation of DEGs with Metabolites
3. Results
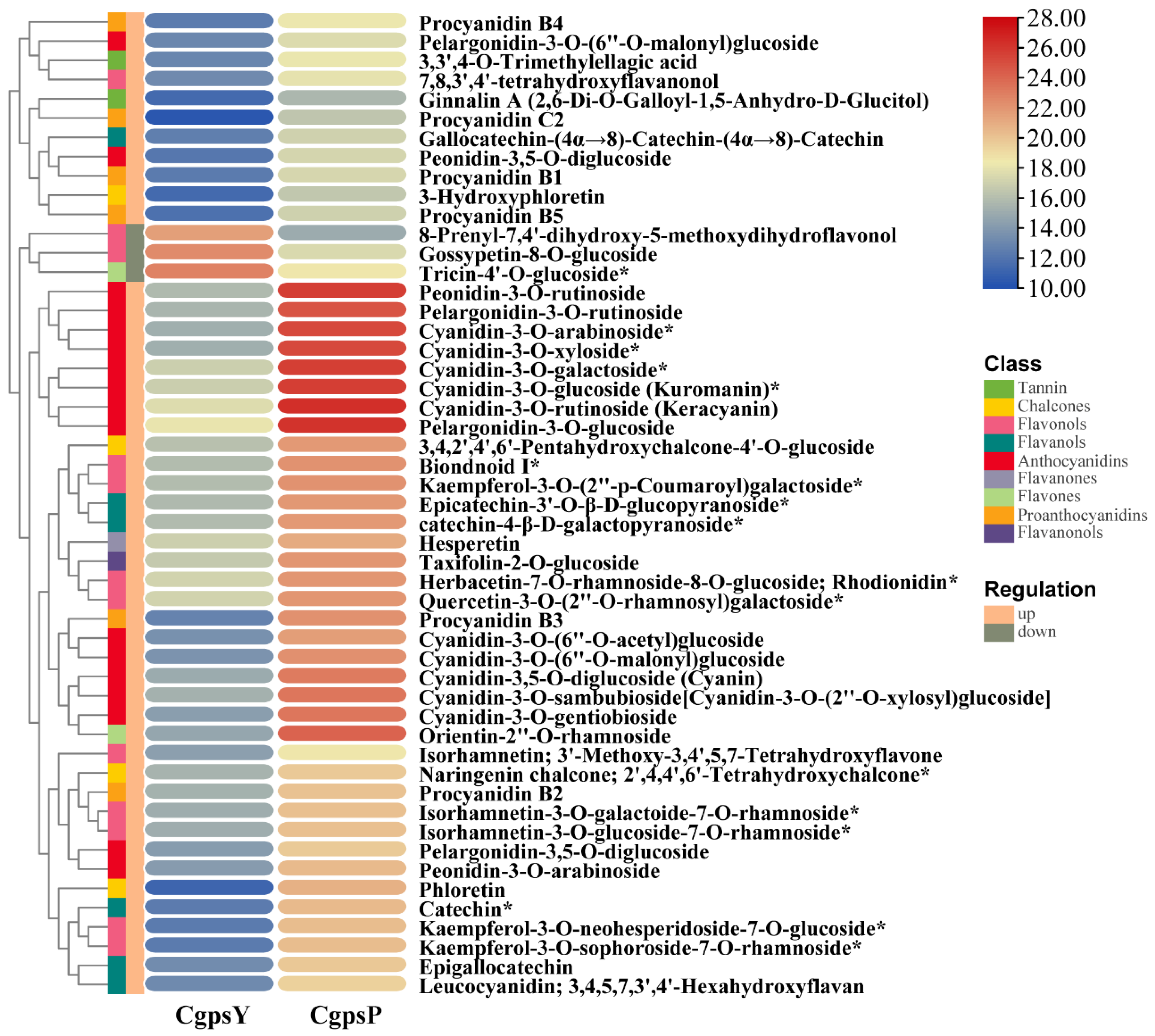
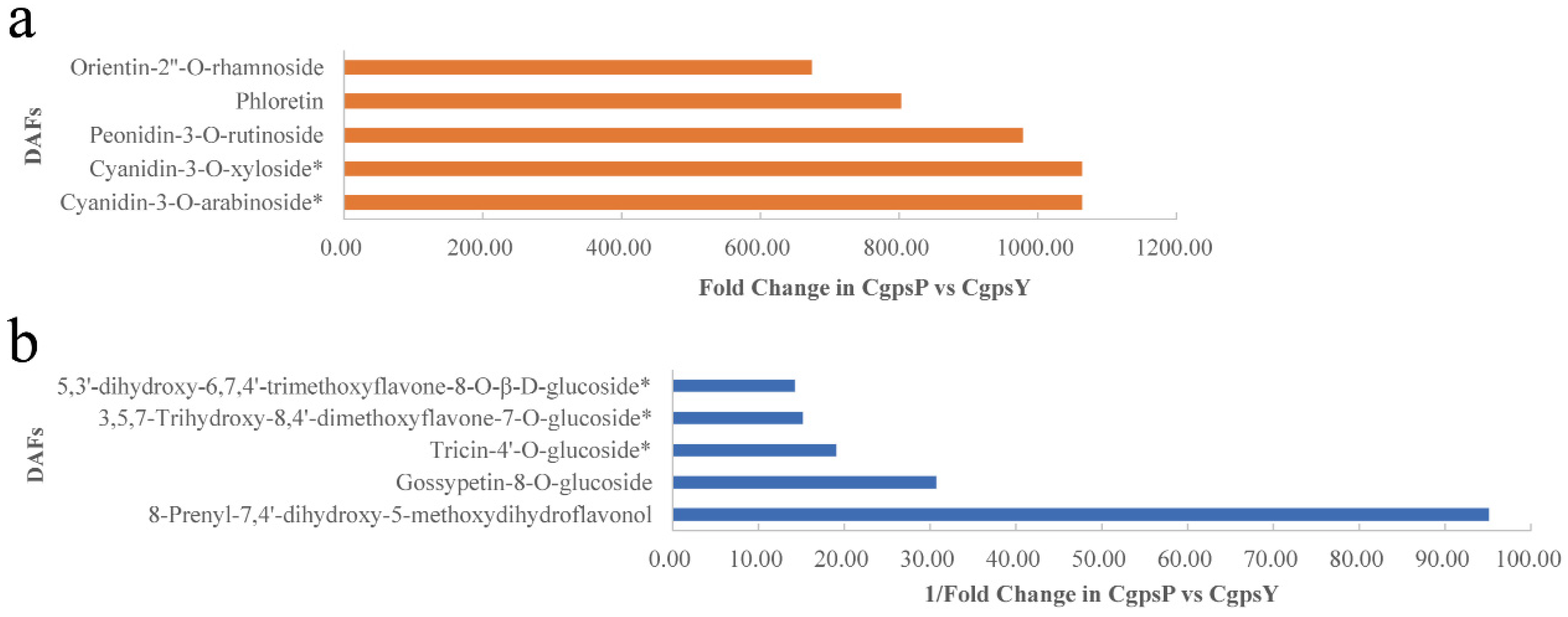
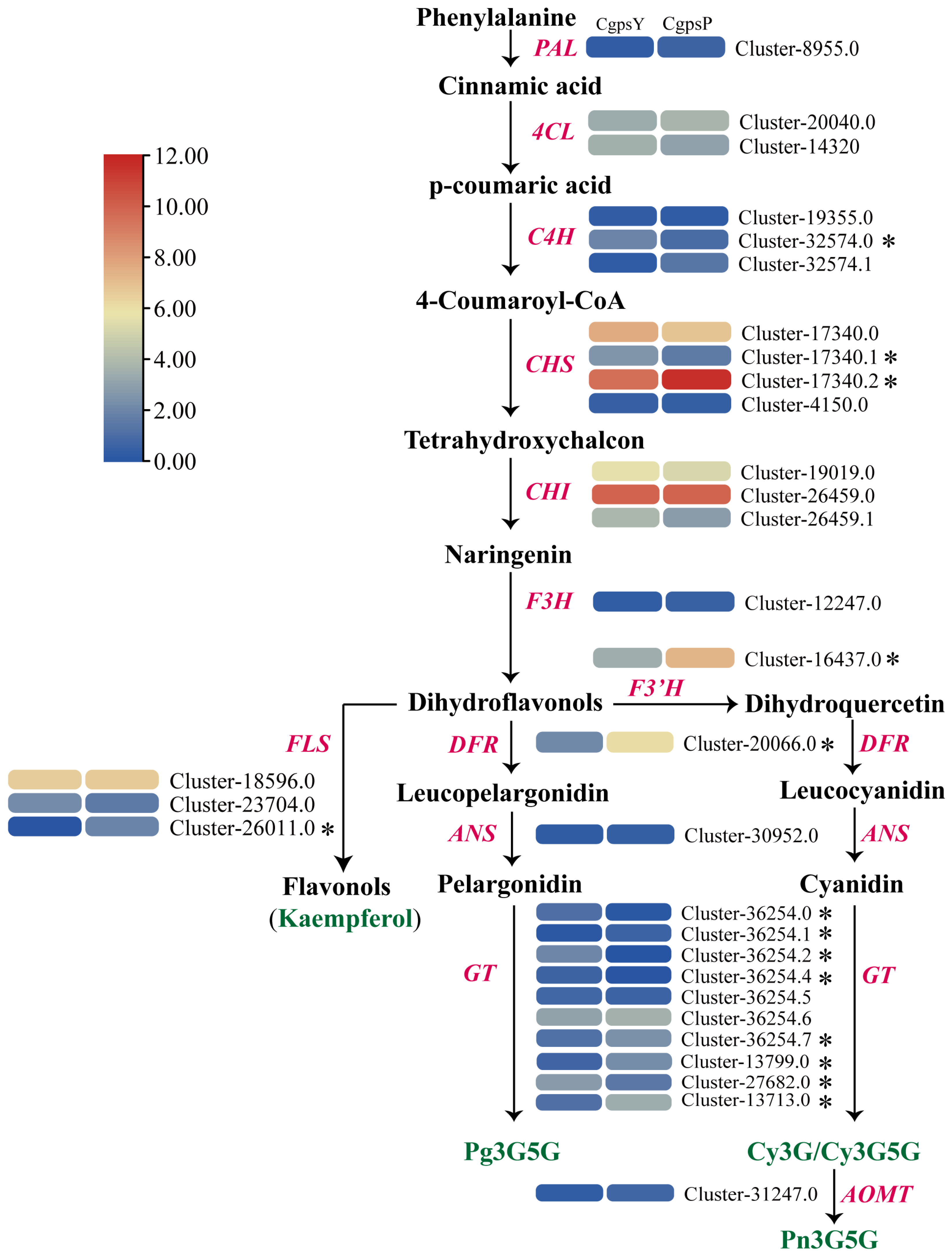
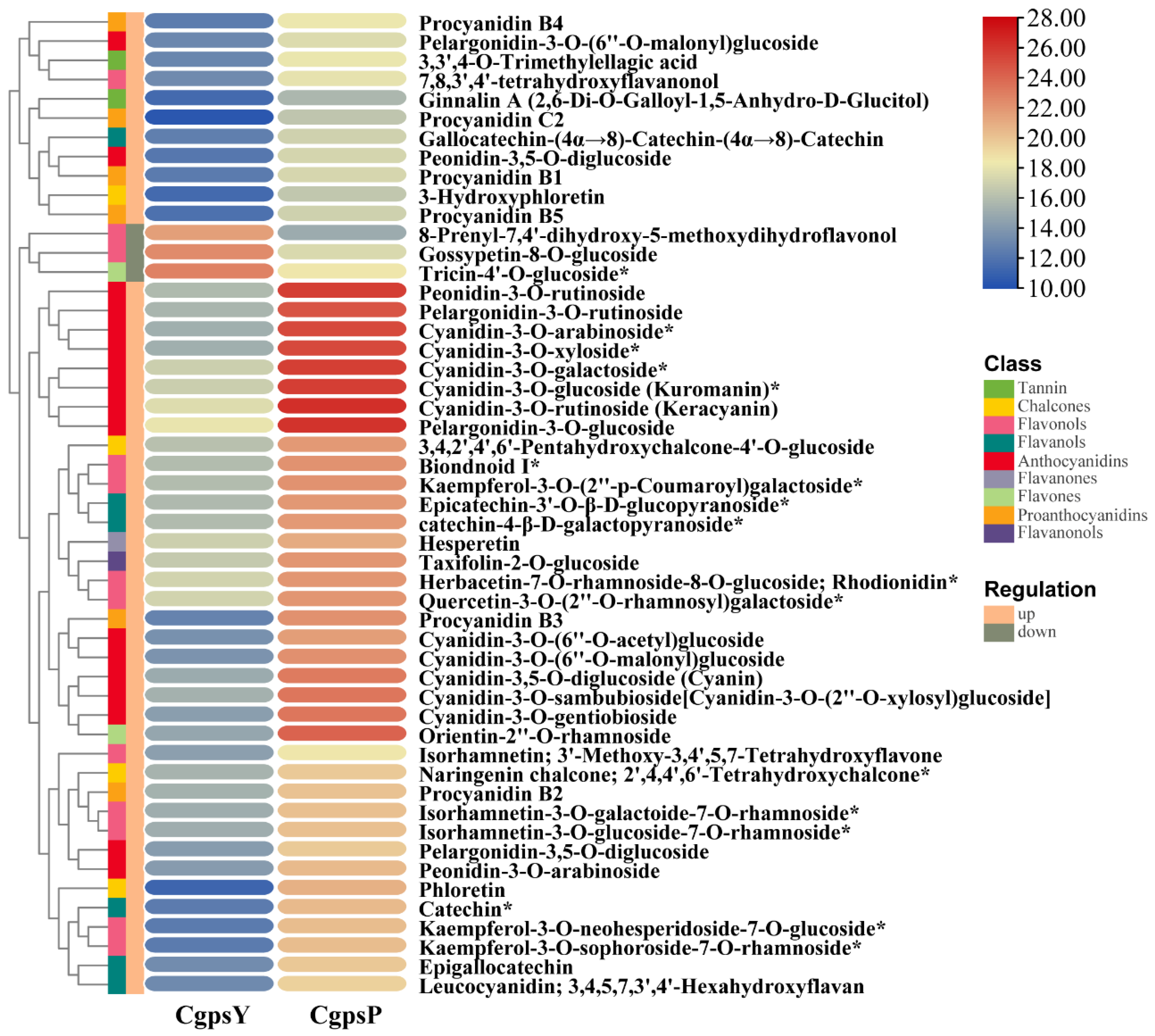
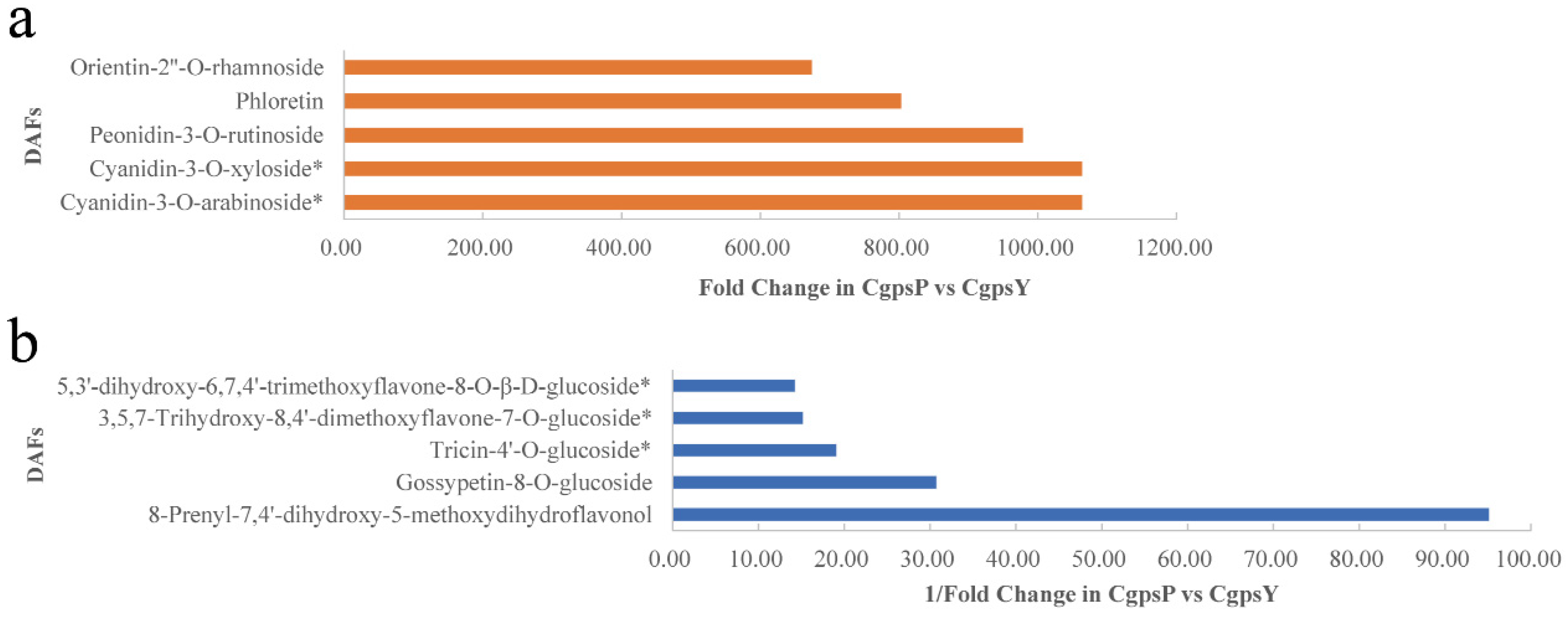
3.1. Flavonoid Metabolic Differences between the Wild Type and the ‘Erebus’ Cultivar of C. glauca
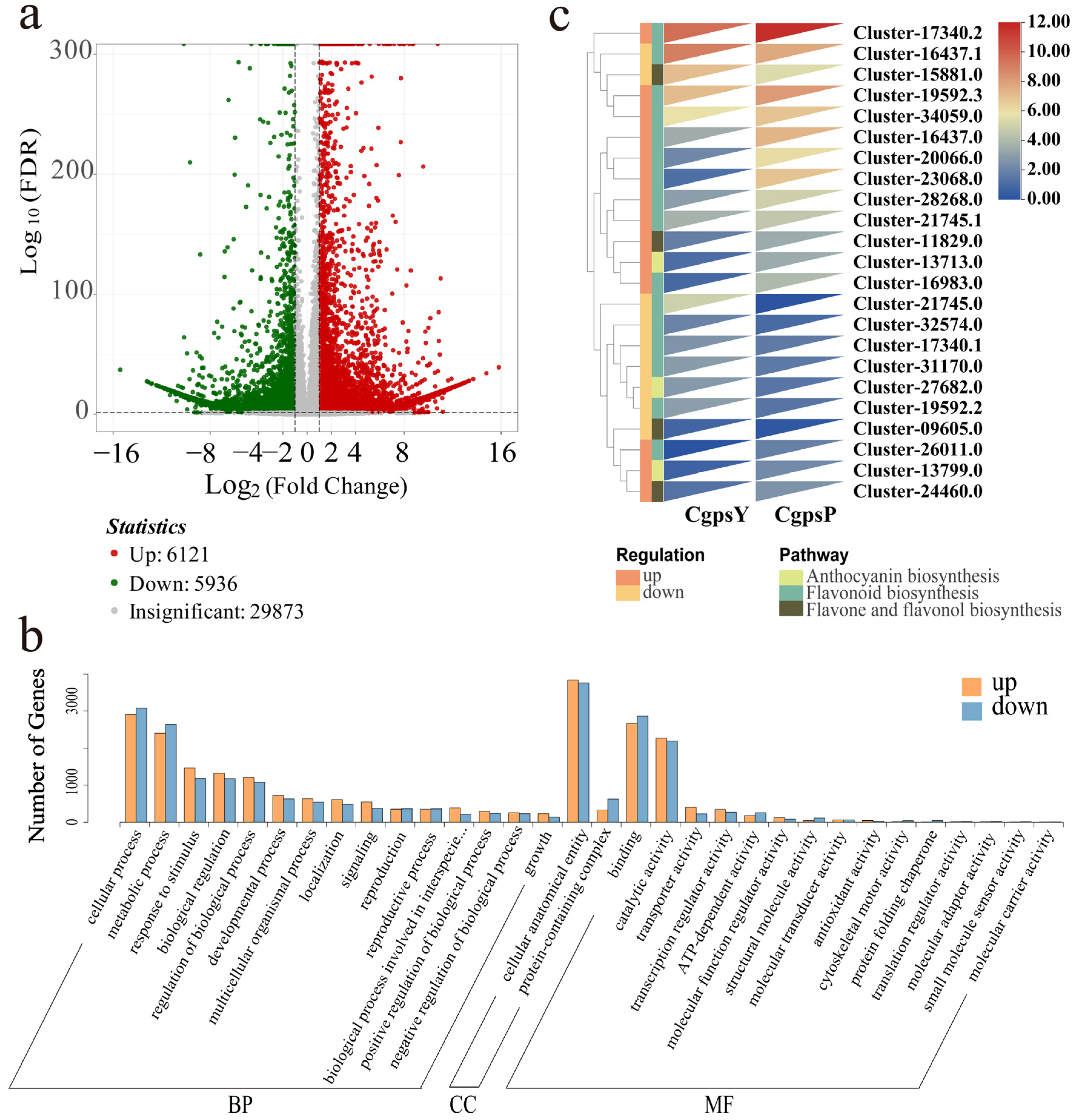
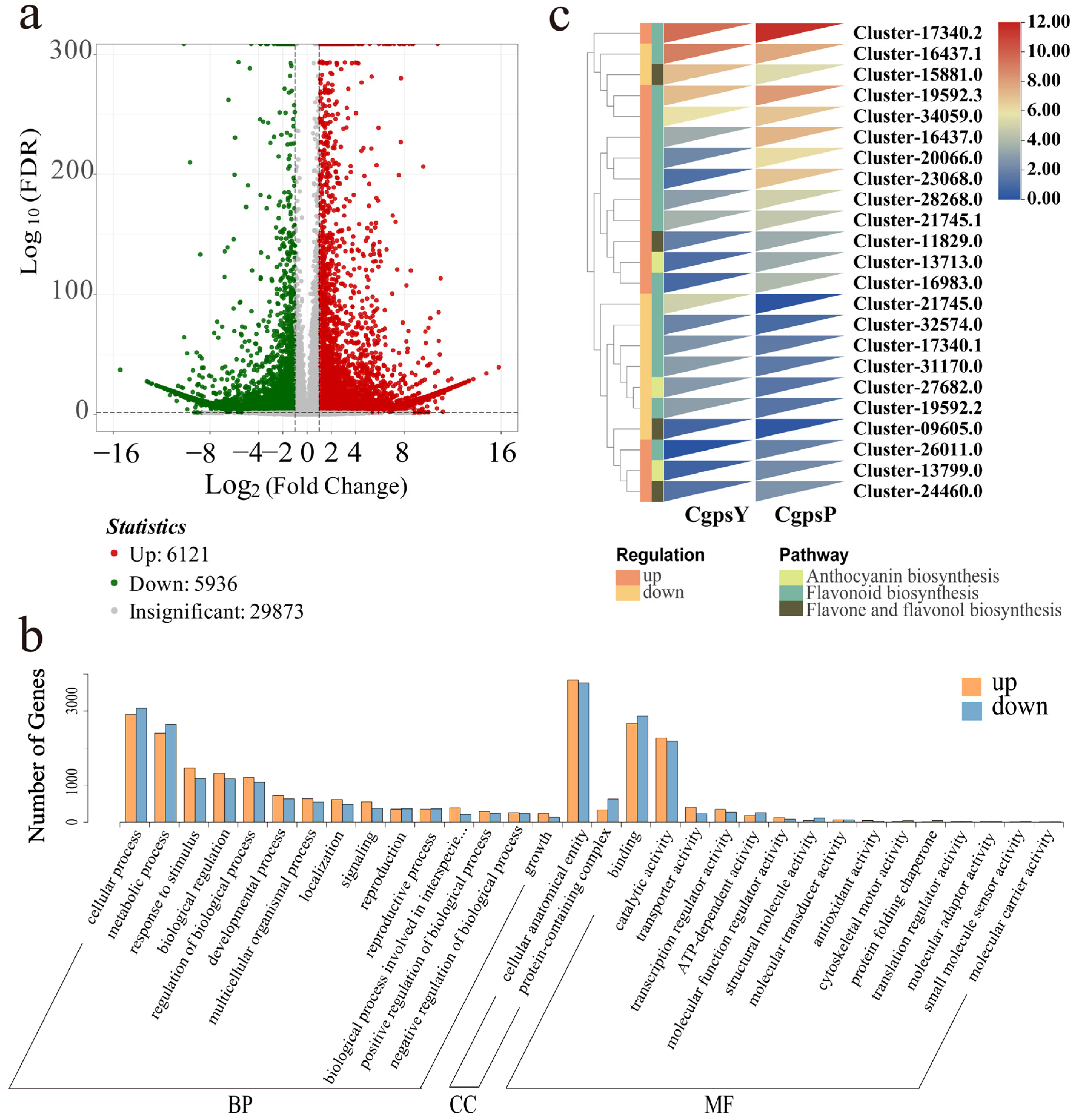
3.2. Transcriptome Overview
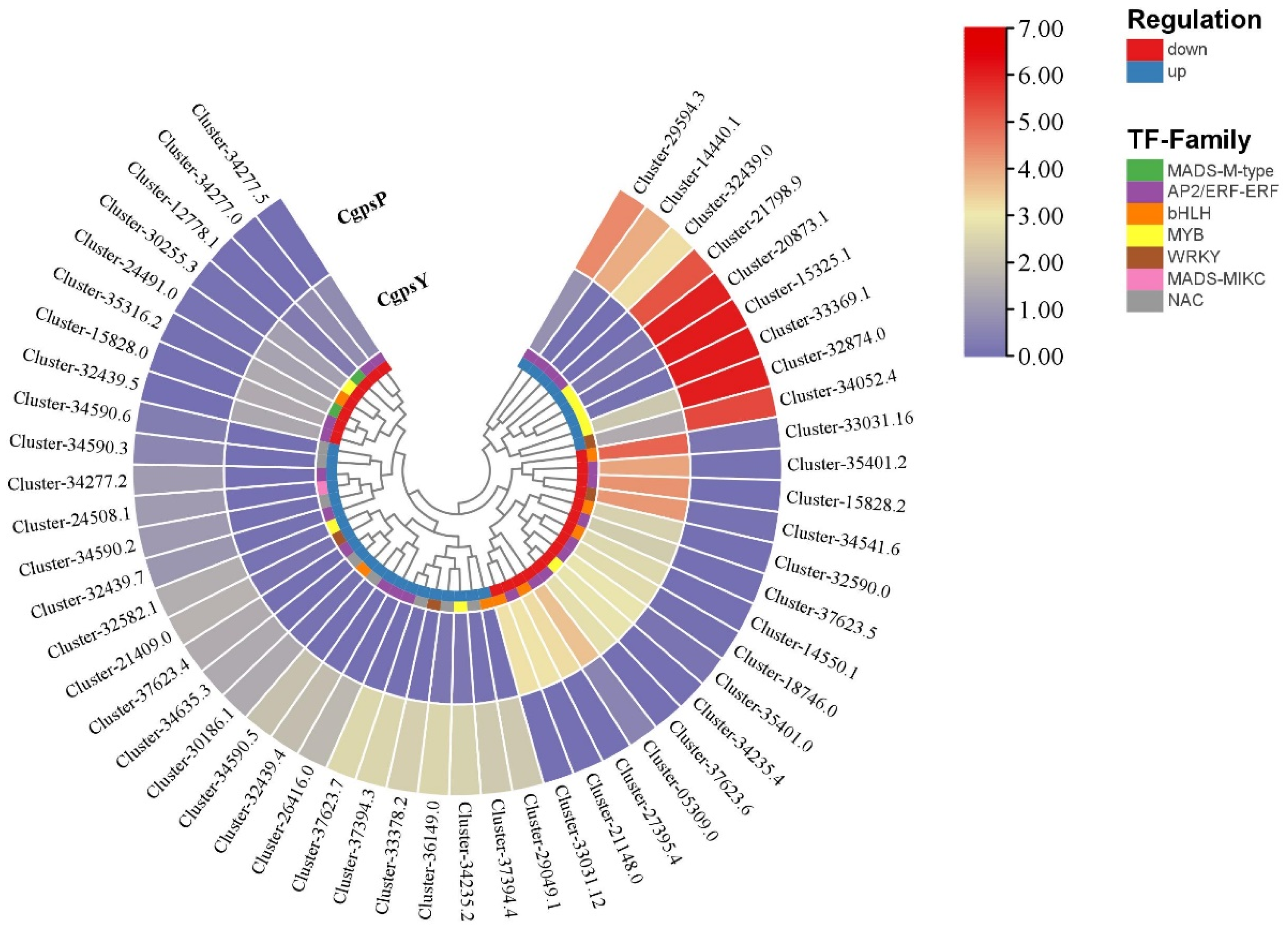
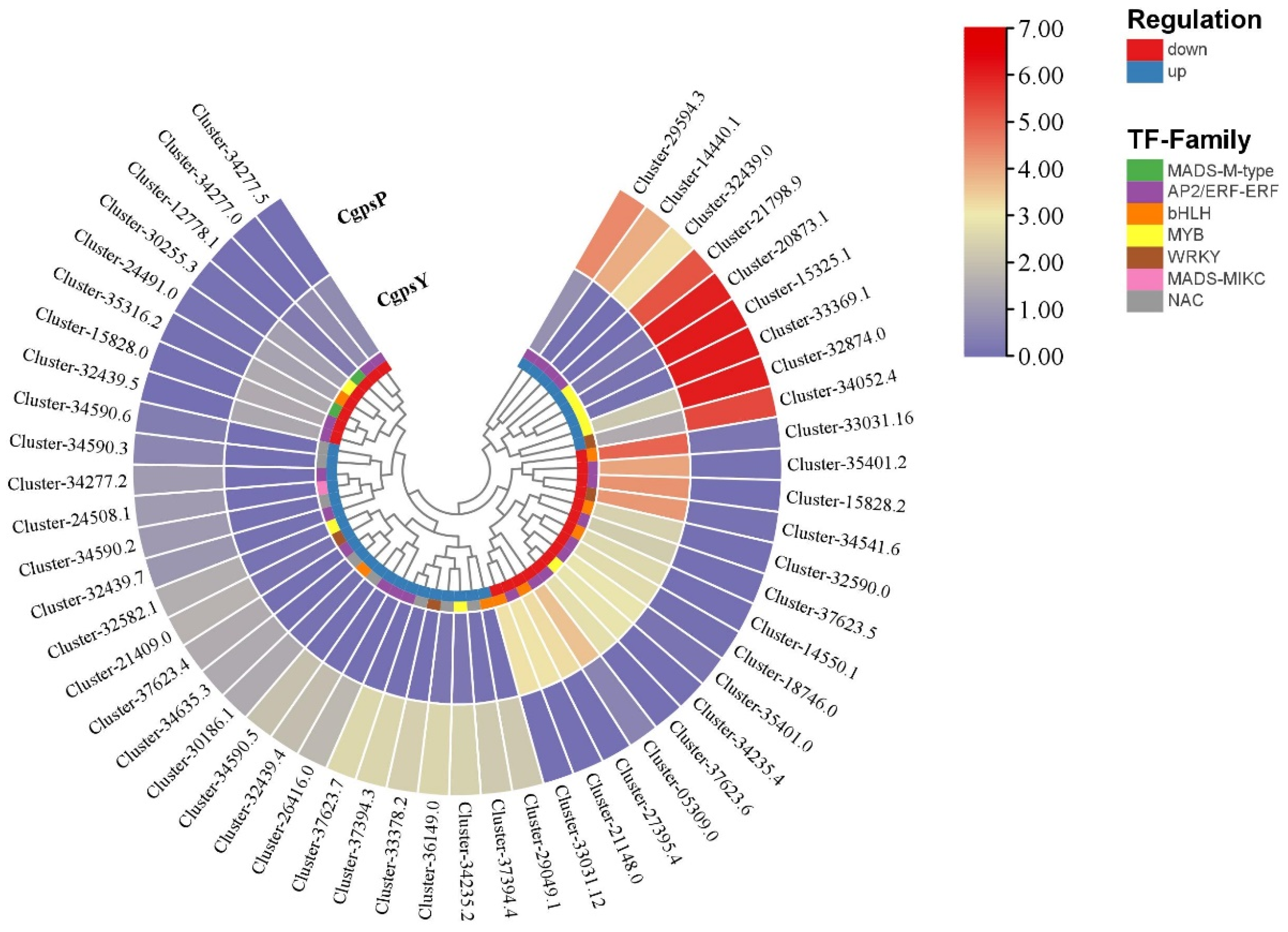
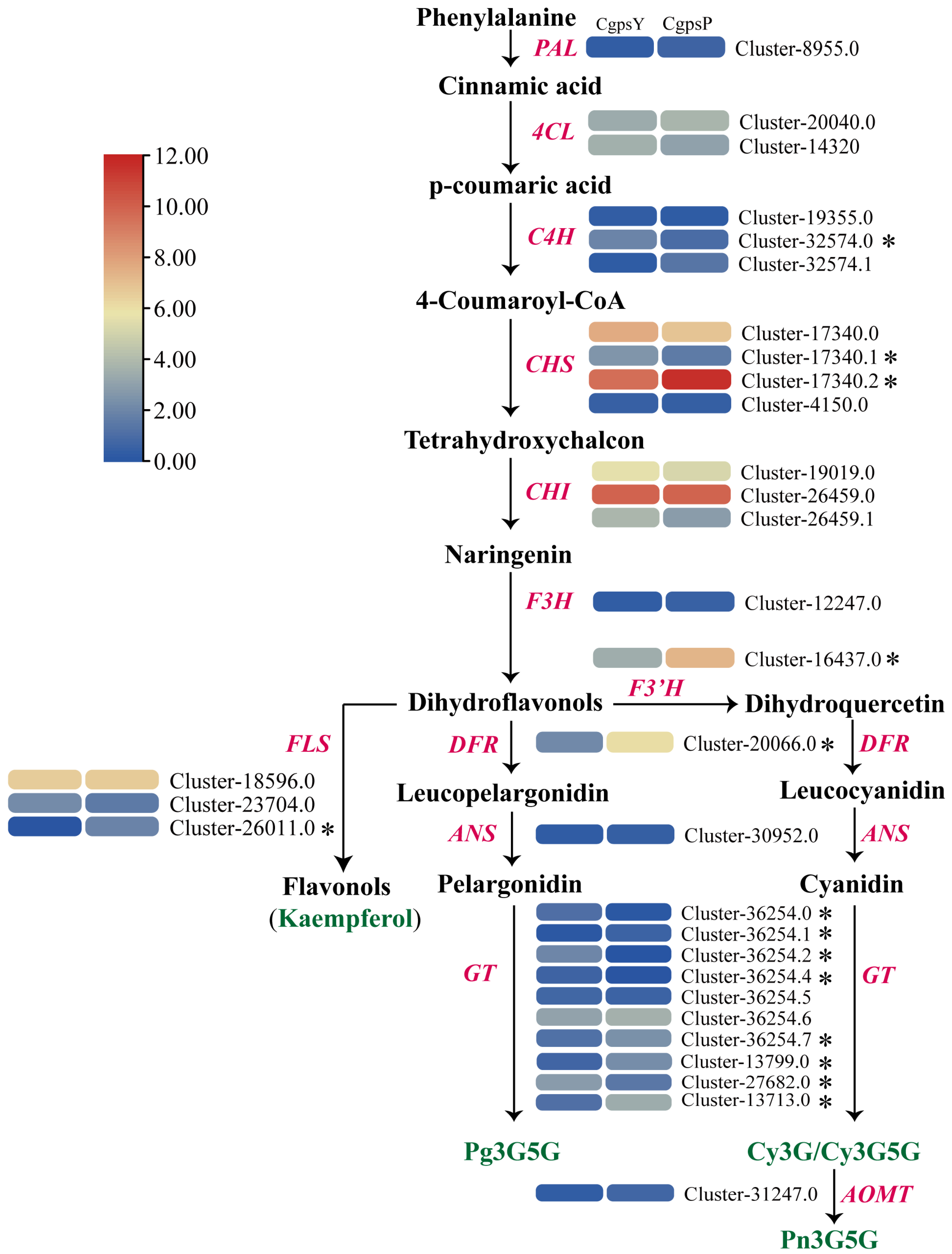
3.3. Differentially Expressed Genes (DEGs) and Transcription Factors Related to Flavonoid Biosynthesis in Petaloid Staminodes of the Wild Type and Cultivar of C. glauca
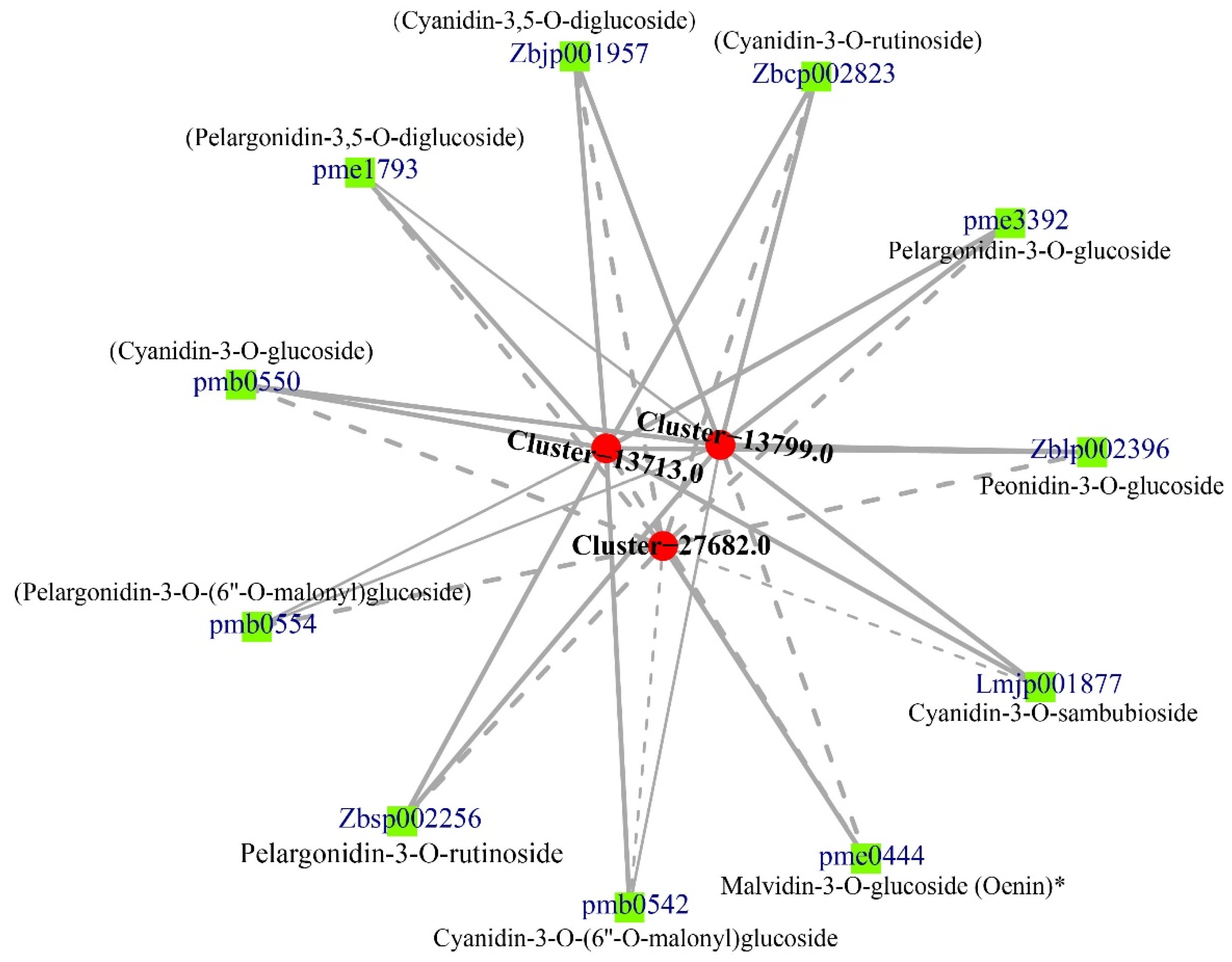
3.4. Correlation between DEGs and Anthocyanins
3.5. Real-Time Quantitative PCR Validation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dahlgren, R.M.T.; Rasmussen, F. Monocotyledon evolution: Characters and phylogenetic estimation. In Evolutionary Biology; Spinger: Boston, MA, USA, 1983; Volume 16, pp. 255–395. [Google Scholar]
- Kirchoff, B.K. Floral ontogeny and evolution in the ginger group of the Zingiberales. In Aspects of Floral Development; Leins, P., Tucker, S.C., Eds.; Cramer: Berlin, Germany, 1988; pp. 45–56. [Google Scholar]
- Kress, W.J. The Phylogeny and classification of the Zingiberales. Ann. Mo. Bot. Gard. 1990, 77, 698–721. [Google Scholar] [CrossRef]
- Kirchoff, B.K. Floral organogenesis in five genera of the Marantaceae and in Canna (Cannaceae). Am. J. Bot. 1983, 70, 508–523. [Google Scholar] [CrossRef]
- Miao, M.; Liu, H.; Kuang, Y.; Zou, P.; Liao, J. Floral vasculature and ontogeny in Canna indica. Nord. J. Bot. 2014, 32, 485–492. [Google Scholar] [CrossRef]
- Tanaka, Y.; Brugliera, F.; Chandler, S. Recent progress of flower colour modification by biotechnology. Int. J. Mol. Sci. 2009, 10, 5350–5369. [Google Scholar] [CrossRef]
- Petroni, K.; Tonelli, C. Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 2011, 181, 219–229. [Google Scholar] [CrossRef]
- Martin, C.; Prescott, A.; Mackay, S.; Bartlett, J.; Vrijlandt, E. Control of anthocyanin biosynthesis in flowers of Antirrhinum majus. Plant J. 1991, 1, 37–49. [Google Scholar] [CrossRef]
- Kazuma, K.; Noda, N.; Suzuki, M. Flavonoid composition related to petal color in different lines of Clitoria ternatea. Phytochemistry 2003, 64, 1133–1139. [Google Scholar] [CrossRef]
- Peng, J.; Dong, X.; Xue, C.; Liu, Z.; Cao, F. Exploring the molecular mechanism of blue flower color formation in Hydrangea macrophylla cv. “Forever Summer”. Front. Plant Sci. 2021, 12, 585665. [Google Scholar] [CrossRef]
- Grotewold, E. The genetics and biochemistry of floral pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780. [Google Scholar] [CrossRef]
- Zhang, Y.; Butelli, E.; Martin, C. Engineering anthocyanin biosynthesis in plants. Curr. Opin. Plant Biol. 2014, 19, 81–90. [Google Scholar] [CrossRef]
- Zhao, D.; Tao, J. Recent advances on the development and regulation of flower color in ornamental plants. Front. Plant Sci. 2015, 6, 261. [Google Scholar] [CrossRef]
- Zhang, H.; Yuan, X.; Wang, R.; Wang, L.; Gao, J.; Wang, H.; Li, Y.; Fu, Z. Comprehensive transcriptome and metabolome characterization of peony ‘Coral Sunset’ petals provides insights into the mechanism of pigment degradation. Horticulturae 2023, 9, 1295. [Google Scholar] [CrossRef]
- Zhao, T.; Yu, Q.; Lin, C.; Liu, H.; Dong, L.; Feng, X.; Liao, J. Analyzing morphology, metabolomics, and transcriptomics offers invaluable insights into the mechanisms of pigment accumulation in the diverse-colored labellum tissues of Alpinia. Plants 2023, 12, 3766. [Google Scholar] [CrossRef]
- Wang, R.; Ren, C.; Dong, S.; Chen, C.; Xian, B.; Wu, Q.; Wang, J.; Pei, J.; Chen, J. Integrated metabolomics and transcriptome analysis of flavonoid biosynthesis in safflower (Carthamus tinctorius L.) with different colors. Front. Plant Sci. 2021, 12, 712038. [Google Scholar] [CrossRef]
- Fu, M.; Yang, X.; Zheng, J.; Wang, L.; Yang, X.; Tu, Y.; Ye, J.; Zhang, W.; Liao, Y.; Cheng, S.; et al. Unraveling the regulatory mechanism of color diversity in Camellia japonica petals by integrative transcriptome and metabolome analysis. Front. Plant Sci. 2021, 12, 685136. [Google Scholar] [CrossRef]
- Nishihara, M.; Nakatsuka, T. Genetic engineering of novel flower colors in floricultural plants: Recent advances via transgenic approaches. Methods Mol. Biol. 2010, 589, 325–347. [Google Scholar]
- Nishihara, M.; Nakatsuka, T. Genetic engineering of flavonoid pigments to modify flower color in floricultural plants. Biotechnol. Lett. 2011, 33, 433–441. [Google Scholar] [CrossRef]
- Jin, X.; Huang, H.; Wang, L.; Sun, Y.; Dai, S. Transcriptomics and metabolite analysis reveals the molecular mechanism of anthocyanin biosynthesis branch pathway in different Senecio cruentus cultivars. Front. Plant Sci. 2016, 7, 1307. [Google Scholar] [CrossRef]
- Xu, L.; Yang, P.; Feng, Y.; Xu, H.; Cao, Y.; Tang, Y.; Yuan, S.; Liu, X.; Ming, J. Spatio temporal transcriptome analysis provides insights into bicolor tepal development in Lilium “Tiny Padhye”. Front. Plant Sci. 2017, 8, 398. [Google Scholar] [CrossRef]
- Li, L.; Ye, J.; Li, H.; Shi, Q. Characterization of metabolites and transcripts involved in flower pigmentation in Primula vulgaris. Front. Plant Sci. 2020, 11, 572517. [Google Scholar] [CrossRef]
- Ma, Y.; Duan, H.; Zhang, F.; Li, Y.; Yang, H.-S.; Tian, F.-P.; Zhou, X.-H.; Wang, C.-M.; Ma, R. Transcriptomic analysis of Lycium ruthenicum Murr. during fruit ripening provides insight into structural and regulatory genes in the anthocyanin biosynthetic pathway. PLoS ONE 2018, 13, e0208627. [Google Scholar] [CrossRef]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 2011, 62, 2465–2483. [Google Scholar] [CrossRef]
- Yin, X.; Lin, X.; Liu, Y.; Irfan, M.; Chen, L.; Zhang, L. Integrated metabolic profiling and transcriptome analysis of pigment accumulation in diverse petal tissues in the lily cultivar ‘Vivian’. BMC Plant Biol. 2020, 20, 446. [Google Scholar] [CrossRef]
- Luo, X.; Sun, D.; Wang, S.; Luo, S.; Fu, Y.; Niu, L.; Shi, Q.; Zhang, Y. Integrating full-length transcriptomics and metabolomics reveals the regulatory mechanisms underlying yellow pigmentation in tree peony (Paeonia suffruticosa Andr.) flowers. Hort. Res. 2021, 1, 008. [Google Scholar]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- He, J.; Jiao, Y. Next-generation sequencing applied to flower development: RNA-seq. In Flower Development: Methods and Protocols; Riechmann, J.L., Wellmer, F., Eds.; Springer: New York, NY, USA, 2014; Volume 1110, pp. 401–411. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Jian, W.; Cao, H.H.; Yuan, S.; Liu, Y.D.; Lu, J.; Lu, W.; Li, N.; Wang, J.; Zou, J.; Tang, N.; et al. SlMYB75, an MYB-type transcription factor, promotes anthocyanin accumulation and enhances volatile aroma production in tomato fruits. Hortic. Res. 2019, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.B.; Li, S.; Zhang, R.F.; Zhao, J.; Chen, Y.; Zhao, Q.; Yao, Y.; You, C.; Zhang, X.; Hao, Y. The bHLH transcription factor MdbHLH3 promotes anthocyanin accumulation and fruit colouration in response to low temperature in apples. Plant Cell Environ. 2012, 35, 1884–1897. [Google Scholar] [PubMed]
- Misyura, M.; Colasanti, J.; Rothstein, S.J. Physiological and genetic analysis of Arabidopsis thaliana anthocyanin biosynthesis mutants under chronic adverse environmental conditions. J. Exp. Bot. 2013, 64, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Tai, D.; Tian, J.; Zhang, J.; Song, T.; Yao, Y. Malus crabapple chalcone synthase gene, McCHS, regulates red petal color and flavonoid biosynthesis. PLoS ONE 2014, 9, 110570. [Google Scholar] [CrossRef] [PubMed]
- Dick, C.; Buenrostro, J.; Butler, T.; Carlson, M.L.; Kliebenstein, D.J.; Whittall, J.B. Arctic mustard flower color polymorphism controlled by petal-specific downregulation at the threshold of the anthocyanin biosynthetic pathway. PLoS ONE 2011, 6, 18230. [Google Scholar] [CrossRef]
- Takos, A.; Jaffé, F.; Jacob, S.; Bogs, J.; Robinson, S.P.; Walker, A.R. Light induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol. 2006, 142, 1216–1232. [Google Scholar] [CrossRef] [PubMed]
- Sunil, L.; Shetty, N.P. Biosynthesis and regulation of anthocyanin pathway genes. Appl. Microbiol. Biot. 2022, 106, 1783–1798. [Google Scholar] [CrossRef] [PubMed]
- Stich, K.; Eidenberger, T.; Wurst, F.; Forkmann, G. Enzymatic conversion of dihydroflavonols to flavan-3, 4-diols using flower extracts of Dianthus caryophyllus L. (carnation). Planta 1992, 187, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Han, Z.; Zhang, J.; Hu, Y.; Song, T.; Yao, Y. The balance of expression of dihydroflavonol 4-reductase and flavonol synthase regulates flavonoid biosynthesis and red foliage coloration in crabapples. Sci. Rep. 2015, 5, 12228. [Google Scholar] [CrossRef]
- Gao, J.; Ren, R.; Wei, Y.; Jin, J.; Ahmad, S.; Lu, C.; Wu, J.; Zheng, C.; Yang, F.; Zhu, G. Comparative metabolomic analysis reveals distinct flavonoid biosynthesis regulation for leaf color development of Cymbidium sinense ‘Red Sun’. Int. J. Mol. Sci. 2020, 21, 1869–1886. [Google Scholar] [CrossRef]
- Noda, N.; Yoshioka, S.; Kishimoto, S.; Nakayama, M.; Douzono, M.; Tanaka, Y.; Aida, R. Generation of blue chrysanthemums by anthocyanin B-ring hydroxylation and glucosylation and its coloration mechanism. Sci. Adv. 2017, 3, e1602785. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wei, G.; Zhou, H.; Gu, C.; Vimolmangkang, S.; Liao, L.; Han, Y. Unraveling the mechanism underlying the glycosylation and methylation of anthocyanins in peach. Plant Physiol. 2014, 166, 1044–1058. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Xu, Z.; Liu, J.; Li, J.; Wang, F.; Xiong, A. Isolation, purification, and characterization of AgUCGalT1, a galactosyltransferase involved in anthocyanin galactosylation in purple celery (Apium graveolens L.). Planta 2018, 247, 1363–1375. [Google Scholar] [CrossRef]
- Xie, L.; Lu, Y.; Zhou, Y.; Hao, X.; Chen, W. Functional analysis of a methyltransferase involved in anthocyanin biosynthesis from blueberries (Vaccinium corymbosum). J. Agric. Food Chem. 2022, 70, 16253–16262. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, T.; Wei, Z.; Liu, H.; Dong, L. Integrated Metabolome and Transcriptome Analyses Reveal That the Flavonoid Metabolic Pathway Is Associated with Pigment Differential Accumulation in Two Colors of Petaloid Staminodes in Canna glauca. Horticulturae 2024, 10, 372. https://doi.org/10.3390/horticulturae10040372
Zhao T, Wei Z, Liu H, Dong L. Integrated Metabolome and Transcriptome Analyses Reveal That the Flavonoid Metabolic Pathway Is Associated with Pigment Differential Accumulation in Two Colors of Petaloid Staminodes in Canna glauca. Horticulturae. 2024; 10(4):372. https://doi.org/10.3390/horticulturae10040372
Chicago/Turabian StyleZhao, Tong, Zehong Wei, Huanfang Liu, and Limei Dong. 2024. "Integrated Metabolome and Transcriptome Analyses Reveal That the Flavonoid Metabolic Pathway Is Associated with Pigment Differential Accumulation in Two Colors of Petaloid Staminodes in Canna glauca" Horticulturae 10, no. 4: 372. https://doi.org/10.3390/horticulturae10040372