
Transcriptional Regulatory Networks Oscillate Seasonal Plasticity of Fruit Metabolism in Melon

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials, Growth Conditions and Sampling
2.2. RNA Extraction and Library Construction
2.3. Metabolomic Analysis
2.4. Transcriptome Analysis
2.5. Machine Learning-Based Co-Expression Analysis
2.6. Enrichment Analysis
3. Results
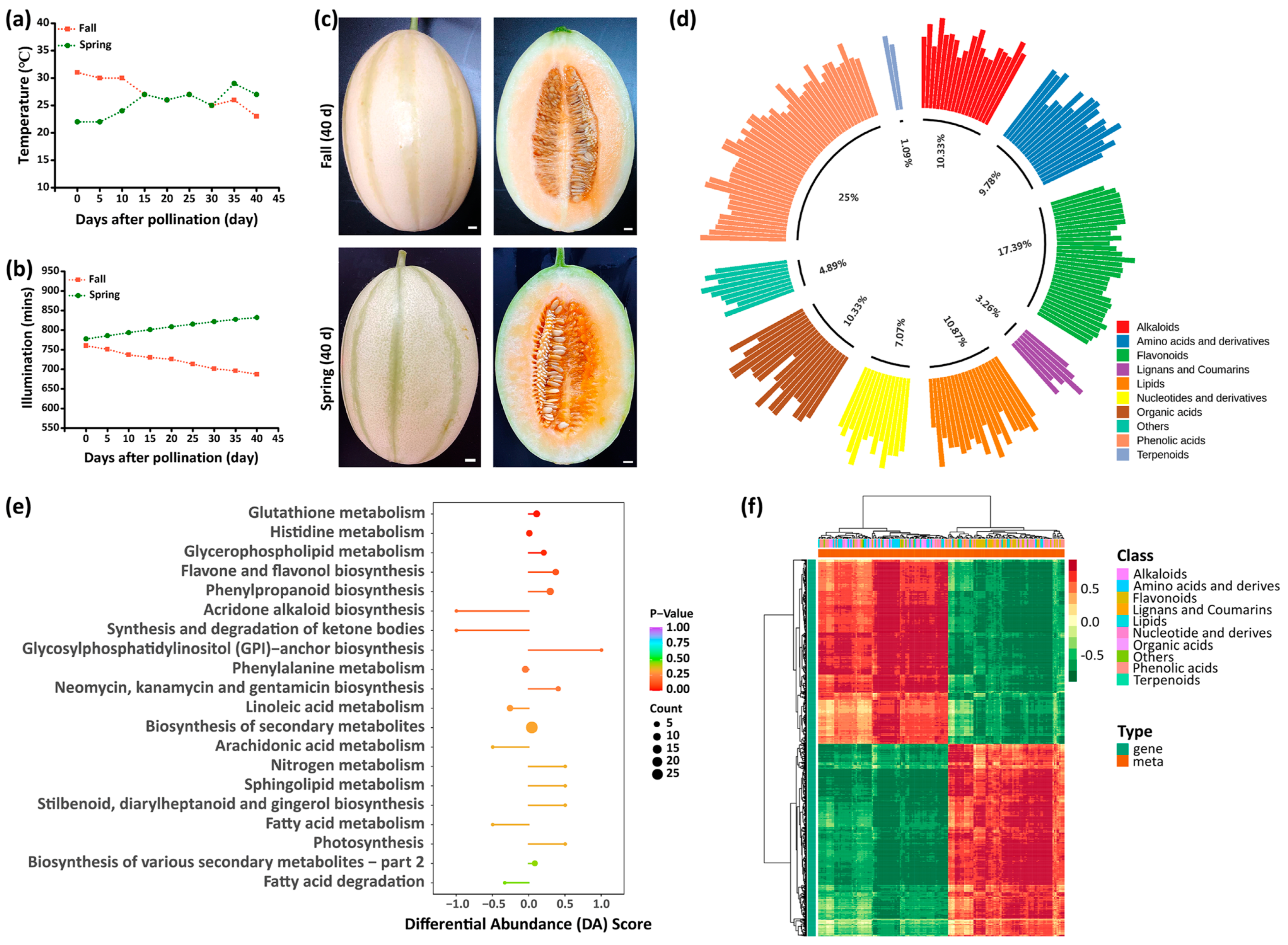
3.1. Metabolomic Analysis of Melons Grown in Spring and Autumn
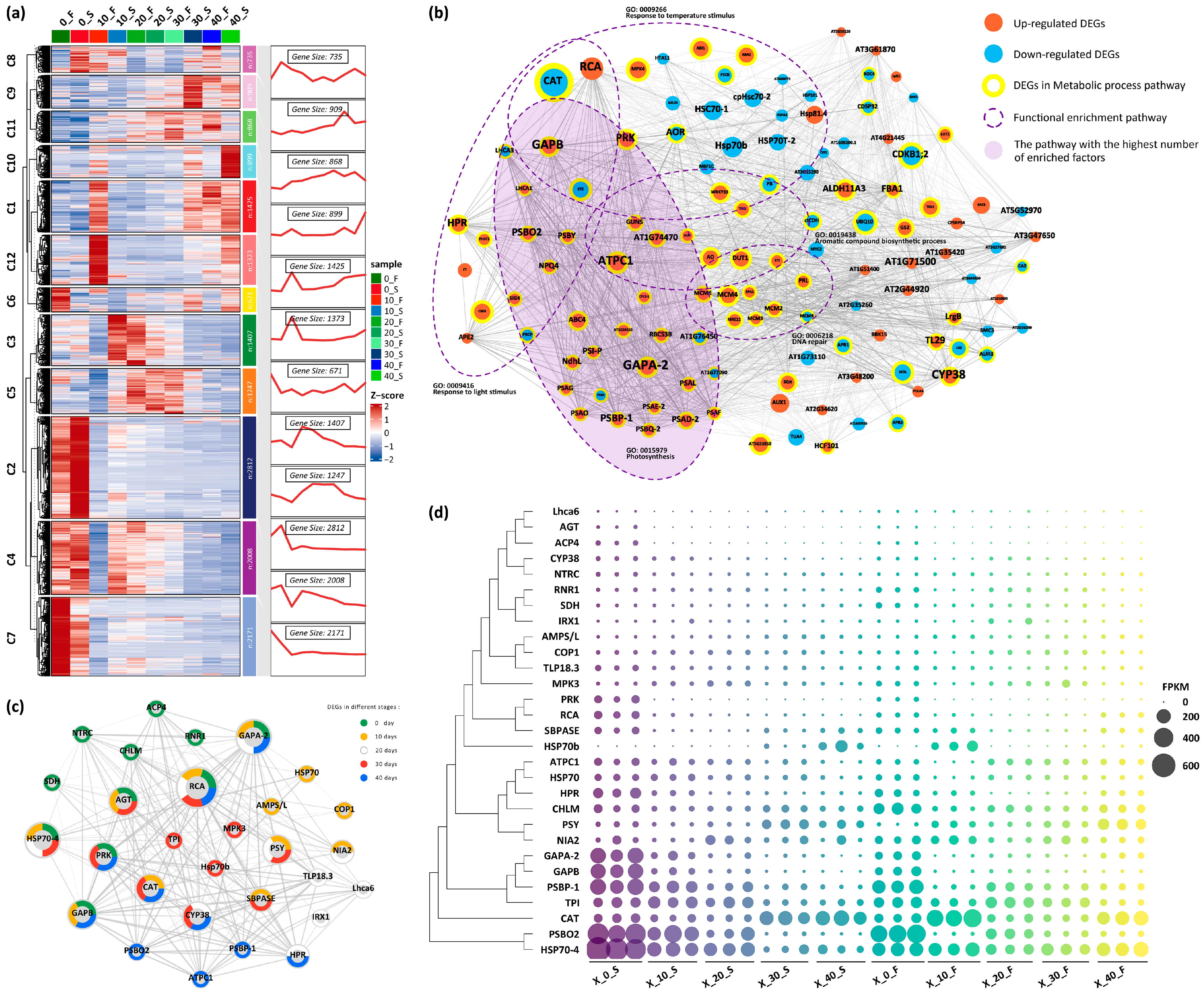
3.2. Transcriptomic Analysis of Melons Grown in Spring and Autumn
3.3. Core Differentially Expressed Genes Were Retained in Spring and Autumn Grown Melons
3.4. Transcriptomic Regulatory Clusters of Spring- and Autumn-Grown Melon Fruits
3.5. Conjoint Analysis of Metabolome and Transcriptome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lester, G.E. Antioxidant, sugar, mineral, and phytonutrient concentrations across edible fruit tissues of orange-fleshed honeydew melon (Cucumis melo L.). J. Agric. Food Chem. 2008, 56, 3694–3698. [Google Scholar] [CrossRef] [PubMed]
- Allwood, J.W.; Cheung, W.; Xu, Y.; Mumm, R.; De Vos, R.C.H.; Deborde, C.; Biais, B.; Maucourt, M.; Berger, Y.; Schaffer, A.A.; et al. Metabolomics in melon: A new opportunity for aroma analysis. Phytochemistry 2014, 99, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Galpaz, N.; Gonda, I.; Shem-Tov, D.; Barad, O.; Tzuri, G.; Lev, S.; Fei, Z.J.; Xu, Y.M.; Mao, L.Y.; Jiao, C.; et al. Deciphering genetic factors that determine melon fruit-quality traits using RNA-Seq-based high-resolution QTL and eQTL mapping. Plant J. 2018, 94, 169–191. [Google Scholar] [CrossRef]
- Moing, A.; Aharoni, A.; Biais, B.; Rogachev, I.; Meir, S.; Brodsky, L.; Allwood, J.W.; Erban, A.; Dunn, W.B.; Kay, L.; et al. Extensive metabolic cross-talk in melon fruit revealed by spatial and developmental combinatorial metabolomics. New Phytol. 2011, 190, 683–696. [Google Scholar] [CrossRef]
- Yang, J.H.; Deng, G.C.; Lian, J.M.; Garraway, J.; Niu, Y.C.; Hu, Z.Y.; Yu, J.Q.; Zhang, M.F. The chromosome-scale genome of melon dissects genetic architecture of important agronomic traits. iScience 2020, 23, 101422. [Google Scholar] [CrossRef] [PubMed]
- Allwood, J.W.; Woznicki, T.L.; Xu, Y.; Foito, A.; Aaby, K.; Sungurtas, J.; Freitag, S.; Goodacre, R.; Stewart, D.; Remberg, S.F.; et al. Application of HPLC-PDA-MS metabolite profiling to investigate the effect of growth temperature and day length on blackcurrant fruit. Metabolomics 2019, 15, 12. [Google Scholar] [CrossRef]
- Almeida, J.; Perez-Fons, L.; Fraser, P.D. A transcriptomic, metabolomic and cellular approach to the physiological adaptation of tomato fruit to high temperature. Plant Cell Environ. 2021, 44, 2211–2229. [Google Scholar] [CrossRef]
- D’Esposito, D.; Ferriello, F.; Dal Molin, A.; Diretto, G.; Sacco, A.; Minio, A.; Barone, A.; Di Monaco, R.; Cavella, S.; Tardella, L.; et al. Unraveling the complexity of transcriptomic, metabolomic and quality environmental response of tomato fruit. BMC Plant Biol. 2017, 17, 66. [Google Scholar] [CrossRef]
- Schwartz, M.D. Phenology: An Integrative Environmental Science; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003. [Google Scholar] [CrossRef]
- Tooke, F.; Battey, N.H. Temperate flowering phenology. J. Exp. Bot. 2010, 61, 2853–2862. [Google Scholar] [CrossRef]
- Kudoh, H. Photoperiod-temperature phase lag: A universal environmental context of seasonal developmental plasticity. Dev. Growth Differ. 2019, 61, 5–11. [Google Scholar] [CrossRef]
- Nagano, A.J.; Kawagoe, T.; Sugisaka, J.; Honjo, M.N.; Iwayama, K.; Kudoh, H. Annual transcriptome dynamics in natural environments reveals plant seasonal adaptation. Nat. Plants 2019, 5, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, H. Molecular phenology in plants: In natura systems biology for the comprehensive understanding of seasonal responses under natural environments. New Phytol. 2016, 210, 399–412. [Google Scholar] [CrossRef]
- Singh, R.K.; Svystun, T.; AlDahmash, B.; Jonsson, A.M.; Bhalerao, R.P. Photoperiod- and temperature-mediated control of phenology in trees—A molecular perspective. New Phytol. 2017, 213, 511–524. [Google Scholar] [CrossRef]
- Monti, L.L.; Bustamante, C.A.; Osorio, S.; Gabilondo, J.; Borsani, J.; Lauxmann, M.A.; Maulion, E.; Valentini, G.; Budde, C.O.; Fernie, A.R.; et al. Metabolic profiling of a range of peach fruit varieties reveals high metabolic diversity and commonalities and differences during ripening. Food Chem. 2016, 190, 879–888. [Google Scholar] [CrossRef]
- Matsuda, F.; Yonekura-Sakakibara, K.; Niida, R.; Kuromori, T.; Shinozaki, K.; Saito, K. MS/MS spectral tag-based annotation of non-targeted profile of plant secondary metabolites. Plant J. 2009, 57, 555–577. [Google Scholar] [CrossRef] [PubMed]
- Rohde, A.; Morreel, K.; Ralph, J.; Goeminne, G.; Hostyn, V.; De Rycke, R.; Kushnir, S.; Van Doorsselaere, J.; Joseleau, J.-P.; Vuylsteke, M.; et al. Molecular Phenotyping of the pal1 and pal2 Mutants of Arabidopsis thaliana Reveals Far-Reaching Consequences on Phenylpropanoid, Amino Acid, and Carbohydrate Metabolism. Plant Cell 2004, 16, 2749–2771. [Google Scholar] [CrossRef]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics-A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Kundariya, H.; Sanchez, R.; Yang, X.D.; Hafner, A.; Mackenzie, S.A. Methylome decoding of RdDM-mediated reprogramming effects in the Arabidopsis MSH1 system. Genome Biol. 2022, 23, 167. [Google Scholar] [CrossRef] [PubMed]
- Gonda, I.; Bar, E.; Portnoy, V.; Lev, S.; Burger, J.; Schaffer, A.A.; Tadmor, Y.A.; Gepstein, S.; Giovannoni, J.J.; Katzir, N.; et al. Branched-chain and aromatic amino acid catabolism into aroma volatiles in Cucumis melo L. fruit. J. Exp. Bot. 2010, 61, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Lau, O.S.; Deng, X.W. The photomorphogenic repressors COP1 and DET1: 20 years later. Trends Plant Sci. 2012, 17, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Schiavon, M.; Pizzeghello, D.; Muscolo, A.; Vaccaro, S.; Francioso, O.; Nardi, S. High Molecular Size Humic Substances Enhance Phenylpropanoid Metabolism in Maize (Zea mays L.). J. Chem. Ecol. 2010, 36, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Burritt, D.J.; Bannister, P. Shoot dry weight, chlorophyll and UV-B-absorbing compounds as indicators of a plant’s sensitivity to UV-B radiation. Ann. Bot. 2000, 86, 1057–1063. [Google Scholar] [CrossRef]
- Chitwood, D.H.; Rundell, S.M.; Li, D.Y.; Woodford, Q.L.; Yu, T.T.; Lopez, J.R.; Greenblatt, D.; Kang, J.; Londo, J.P. Climate and Developmental Plasticity: Interannual Variability in Grapevine Leaf Morphology. Plant Physiol. 2016, 170, 1480–1491. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. Mitogen-activated protein kinase cascades in plant signaling. J. Integr. Plant Biol. 2022, 64, 301–341. [Google Scholar] [CrossRef]
- Dixon, R.A.; Paiva, N.L. Stress-Induced Phenylpropanoid Metabolism. Plant Cell 1995, 7, 1085–1097. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid Biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef]
- Xing, L.B.; Li, Y.M.; Qi, S.Y.; Zhang, C.G.; Ma, W.C.; Zuo, X.Y.; Liang, J.Y.; Gao, C.; Jia, P.; Shah, K.; et al. Comparative RNA-Sequencing and DNA Methylation Analyses of Apple (Malus domestica Borkh.) Buds with Diverse Flowering Capabilities Reveal Novel Insights into the Regulatory Mechanisms of Flower Bud Formation. Plant Cell Physiol. 2019, 60, 1702–1721. [Google Scholar] [CrossRef]
- Shi, J.Y.; Du, X.G. Transcriptome analysis reveals the regulation of cyclic nucleotide-gated ion channels in response to exogenous abscisic acid and calcium treatment under drought stress in tomato. Front. Genet. 2023, 14, 1139087. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Campos, E.; García-Villaraco, A.; Montero-Palmero, M.B.; Gutiérrez-Mañero, F.J.; Ramos-Solano, B. Bacillus G7 improves adaptation to salt stress in Olea europaea L. plantlets, enhancing water use efficiency and preventing oxidative stress. Sci. Rep. 2023, 13, 22507. [Google Scholar] [CrossRef]
- Sun, X.X.; Feng, D.L.; Liu, M.Y.; Qin, R.X.; Li, Y.; Lu, Y.; Zhang, X.M.; Wang, Y.H.; Shen, S.X.; Ma, W.; et al. Single-cell transcriptome reveals dominant subgenome expression and transcriptional response to heat stress in Chinese cabbage. Genome Biol. 2022, 23, 262. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.L.; Chen, B.H.; Chen, X.J.; Guo, Y.Y.; Yang, H.L.; Li, X.Z.; Wang, G.Y. Transcriptome profiling of pumpkin (Cucurbita moschata Duch.) leaves infected with powdery mildew. PLoS ONE 2018, 13, e0190175. [Google Scholar] [CrossRef] [PubMed]
- Fraser, C.M.; Chapple, C. The phenylpropanoid pathway in Arabidopsis. Arab. Book 2011, 9, e0152. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.; Caparros-Ruiz, D.; Grotewold, E. Grass phenylpropanoids: Regulate before using! Plant Sci. 2012, 184, 112–120. [Google Scholar] [CrossRef]
- Liu, J.; Osbourn, A.; Ma, P. MYB Transcription Factors as Regulators of Phenylpropanoid Metabolism in Plants. Mol. Plant 2015, 8, 689–708. [Google Scholar] [CrossRef]
- Lanot, A.; Hodge, D.; Lim, E.-K.; Vaistij, F.E.; Bowles, D.J. Redirection of flux through the phenylpropanoid pathway by increased glucosylation of soluble intermediates. Planta 2008, 228, 609–616. [Google Scholar] [CrossRef]
- Chen, X.; Ding, Y.; Yang, Y.; Song, C.; Wang, B.; Yang, S.; Guo, Y.; Gong, Z. Protein kinases in plant responses to drought, salt, and cold stress. J. Integr. Plant Biol. 2021, 63, 53–78. [Google Scholar] [CrossRef]
- Fujita, M.; Fujita, Y.; Noutoshi, Y.; Takahashi, F.; Narusaka, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Crosstalk between abiotic and biotic stress responses: A current view from the points of convergence in the stress signaling networks. Curr. Opin. Plant Biol. 2006, 9, 436–442. [Google Scholar] [CrossRef]
- Gong, Z.; Xiong, L.; Shi, H.; Yang, S.; Herrera-Estrella, L.R.; Xu, G.; Chao, D.-Y.; Li, J.; Wang, P.-Y.; Qin, F.; et al. Plant abiotic stress response and nutrient use efficiency. Sci. China-Life Sci. 2020, 63, 635–674. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Falcone Ferreyra, M.L.; Rius, S.P.; Casati, P. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 2012, 3, 222. [Google Scholar] [CrossRef] [PubMed]
- Zoratti, L.; Karppinen, K.; Escobar, A.L.; Haggman, H.; Jaakola, L. Light-controlled flavonoid biosynthesis in fruits. Front. Plant Sci. 2014, 5, 534. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, S. Mitogen-activated protein kinase cascades in signaling plant growth and development. Trends Plant Sci. 2015, 20, 56–64. [Google Scholar] [CrossRef]
- Zhang, M.; Su, J.; Zhang, Y.; Xu, J.; Zhang, S. Conveying endogenous and exogenous signals: MAPK cascades in plant growth and defense. Curr. Opin. Plant Biol. 2018, 45, 1–10. [Google Scholar] [CrossRef]
- Chayut, N.; Yuan, H.; Saar, Y.; Zheng, Y.; Sun, T.; Zhou, X.; Hermanns, A.; Oren, E.; Faigenboim, A.; Hui, M.; et al. Comparative transcriptome analyses shed light on carotenoid production and plastid development in melon fruit. Hortic. Res. 2021, 8, 112. [Google Scholar] [CrossRef]
- Diao, Q.; Tian, S.; Cao, Y.; Yao, D.; Fan, H.; Zhang, Y. Transcriptome analysis reveals association of carotenoid metabolism pathway with fruit color in melon. Sci. Rep. 2023, 13, 5004. [Google Scholar] [CrossRef]
- Zhang, A.; Zheng, J.; Chen, X.; Shi, X.; Wang, H.; Fu, Q. Comprehensive Analysis of Transcriptome and Metabolome Reveals the Flavonoid Metabolic Pathway Is Associated with Fruit Peel Coloration of Melon. Molecules 2021, 26, 2830. [Google Scholar] [CrossRef]
- Cohen, H.; Dong, Y.; Szymanski, J.; Lashbrooke, J.; Meir, S.; Almekias-Siegl, E.; Zeisler-Diehl, V.V.; Schreiber, L.; Aharoni, A. A Multilevel Study of Melon Fruit Reticulation Provides Insight into Skin Ligno-Suberization Hallmarks. Plant Physiol. 2019, 179, 1486–1501. [Google Scholar] [CrossRef]
- Usman, M.G.; Rafii, M.Y.; Martini, M.Y.; Yusuff, O.A.; Ismail, M.R.; Miah, G. Molecular analysis of Hsp70 mechanisms in plants and their function in response to stress. Biotechnol. Genet. Eng. Rev. 2017, 33, 26–39. [Google Scholar] [CrossRef]
- Adaskaveg, J.A.; Blanco-Ulate, B. Targeting ripening regulators to develop fruit with high quality and extended shelf life. Curr. Opin. Biotechnol. 2023, 79, 102872. [Google Scholar] [CrossRef]
- Tadmor, Y.; Burger, J.; Yaakov, I.; Feder, A.; Libhaber, S.E.; Portnoy, V.; Meir, A.; Tzuri, G.; Saar, U.; Rogachev, I.; et al. Genetics of Flavonoid, Carotenoid, and Chlorophyll Pigments in Melon Fruit Rinds. J. Agric. Food Chem. 2010, 58, 10722–10728. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, L.; Rodriguez-Concepcion, M. Manipulation of plastidial protein quality control components as a new strategy to improve carotenoid contents in tomato fruit. Front. Plant Sci. 2019, 10, 1071. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Huang, X.; Deng, X.W. The photomorphogenic central repressor COP1: Conservation and functional diversification during evolution. Plant Commun. 2020, 1, 100044. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Muqarab, R.; Waseem, M. The Solanum melongena COP1 delays fruit ripening and influences ethylene signaling in tomato. J. Plant Physiol. 2019, 240, 152997. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Piao, R.H.; Lee, G.; Koh, E.; Lee, Y.; Woo, S.; Reflinur; Jiang, W.Z.; Septiningsih, E.M.; Thomson, M.J.; et al. OsCOP1 regulates embryo development and flavonoid biosynthesis in rice (Oryza sativa L.). Theor. Appl. Genet. 2021, 134, 2587–2601. [Google Scholar] [CrossRef]
- Bhatia, C.; Gaddam, S.R.; Pandey, A.; Trivedi, P.K. COP1 mediates light-dependent regulation of flavonol biosynthesis through HY5 in Arabidopsis. Plant Sci. 2021, 303, 110760. [Google Scholar] [CrossRef]
- Bhatia, C.; Pandey, A.; Gaddam, S.R.; Hoecker, U.; Trivedi, P.K. Low temperature-enhanced flavonol synthesis requires light-associated regulatory components in Arabidopsis thaliana. Plant Cell Physiol. 2018, 59, 2099–2112. [Google Scholar] [CrossRef]
- Andralojc, P.J.; Carmo-Silva, E.; Degen, G.E.; Parry, M.A.J. Increasing metabolic potential: C-fixation. Chloroplasts Capture Prod. Modul. Plants 2018, 62, 109–118. [Google Scholar] [CrossRef]
- Flecken, M.; Wang, H.P.; Popilka, L.; Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Dual Functions of a Rubisco Activase in Metabolic Repair and Recruitment to Carboxysomes. Cell 2020, 183, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Degen, G.E.; Orr, D.J.; Carmo-Silva, E. Heat-induced changes in the abundance of wheat Rubisco activase isoforms. New Phytol. 2021, 229, 1298–1311. [Google Scholar] [CrossRef] [PubMed]
- Degen, G.E.; Worrall, D.; Carmo-Silva, E. An isoleucine residue acts as a thermal and regulatory switch in wheat Rubisco activase. Plant J. 2020, 103, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.C.; Sakoda, K.; Fukayama, H.; Kondo, E.; Suzuki, Y.; Makino, A.; Terashima, I.; Yamori, W. Overexpression of both Rubisco and Rubisco activase rescues rice photosynthesis and biomass under heat stress. Plant Cell Environ. 2021, 44, 2308–2320. [Google Scholar] [CrossRef]
- Wijewardene, I.; Mishra, N.; Sun, L.; Smith, J.; Zhu, X.L.; Payton, P.; Shen, G.X.; Zhang, H. Improving drought-, salinity-, and heat-tolerance in transgenic plants by co-overexpressing Arabidopsis vacuolar pyrophosphatase gene AVP1 and Larrea Rubisco activase gene RCA. Plant Sci. 2020, 296, 110499. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Z.; Zhang, J.; Yang, X.; Deng, G.; Sun, J.; Xia, Y.; Hu, Z.; Zhang, M.; Yang, J. Transcriptional Regulatory Networks Oscillate Seasonal Plasticity of Fruit Metabolism in Melon. Horticulturae 2024, 10, 993. https://doi.org/10.3390/horticulturae10090993
Gong Z, Zhang J, Yang X, Deng G, Sun J, Xia Y, Hu Z, Zhang M, Yang J. Transcriptional Regulatory Networks Oscillate Seasonal Plasticity of Fruit Metabolism in Melon. Horticulturae. 2024; 10(9):993. https://doi.org/10.3390/horticulturae10090993
Chicago/Turabian StyleGong, Zihui, Jiejing Zhang, Xiaodong Yang, Guancong Deng, Ji Sun, Yuelin Xia, Zhongyuan Hu, Mingfang Zhang, and Jinghua Yang. 2024. "Transcriptional Regulatory Networks Oscillate Seasonal Plasticity of Fruit Metabolism in Melon" Horticulturae 10, no. 9: 993. https://doi.org/10.3390/horticulturae10090993
APA StyleGong, Z., Zhang, J., Yang, X., Deng, G., Sun, J., Xia, Y., Hu, Z., Zhang, M., & Yang, J. (2024). Transcriptional Regulatory Networks Oscillate Seasonal Plasticity of Fruit Metabolism in Melon. Horticulturae, 10(9), 993. https://doi.org/10.3390/horticulturae10090993