
Genome-Wide Identification of miRNAs in Oily Persimmon (Diospyros oleifera Cheng) and Their Functional Targets Associated with Proanthocyanidin Metabolism

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Date Collection for the Prediction of miRNAs and Pre-miRNAs in Persimmon
2.2. Phylogenetic and Conservation Analysis of the miRNAs
2.3. Predicting the Target Genes of the miRNAs
2.4. Genome Location of the miRNAs in D. oleifera
2.5. RNA Extraction and cDNA Synthesis
2.6. RT-qPCR Analysis of the Pre-miRNAs and Their Target Genes
2.7. Predicting the Cis-Acting Elements in the Promoter Region of the Pre-miRNA Genes
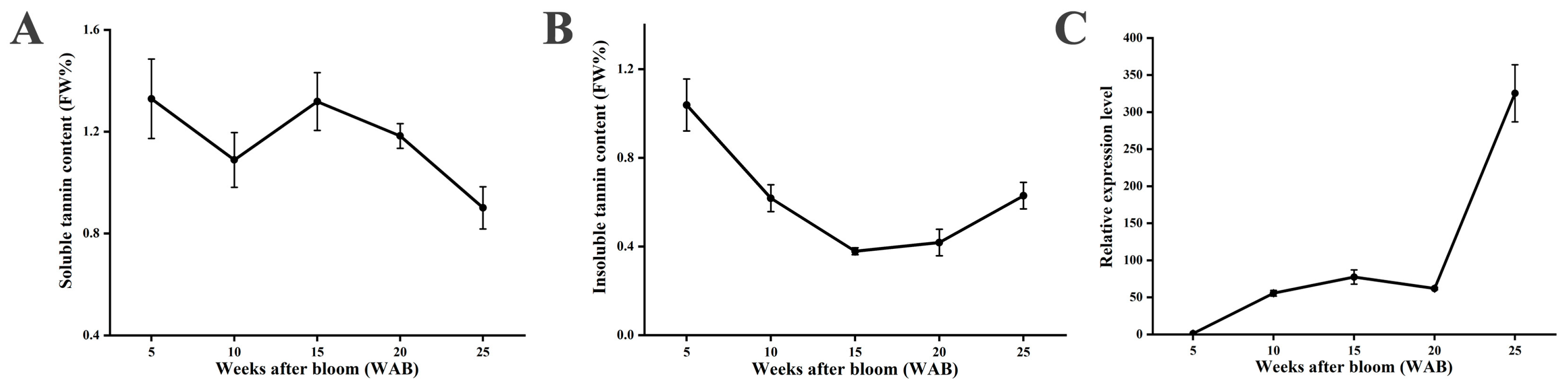
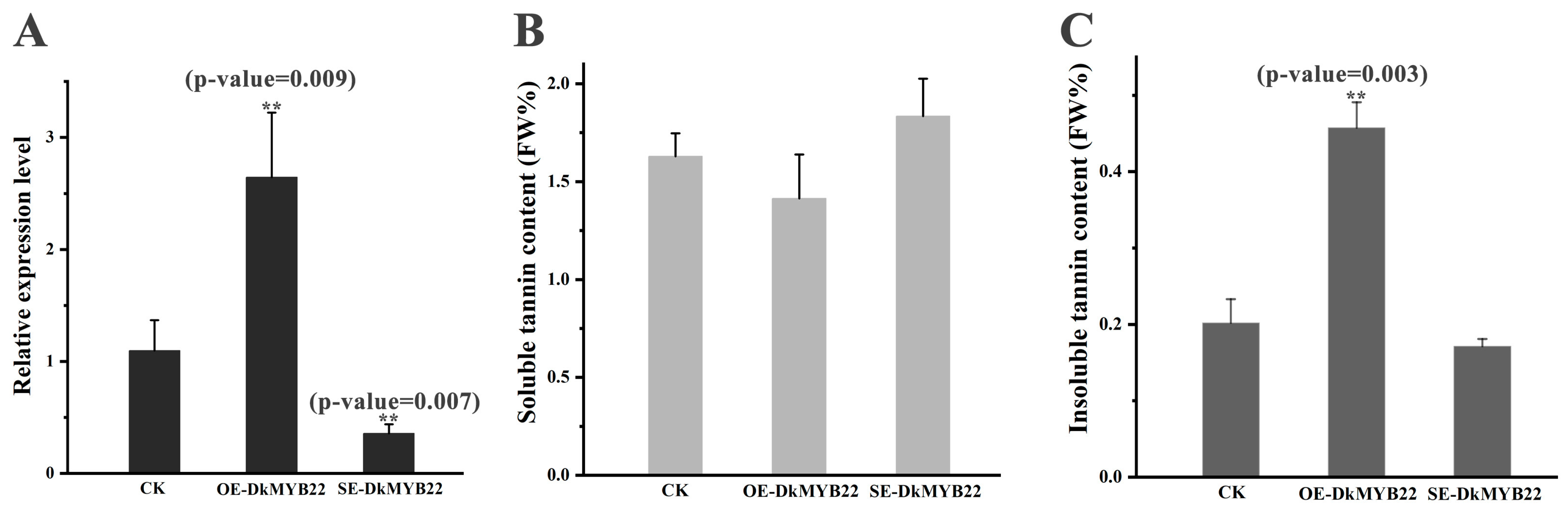
2.8. PA Content Measurement
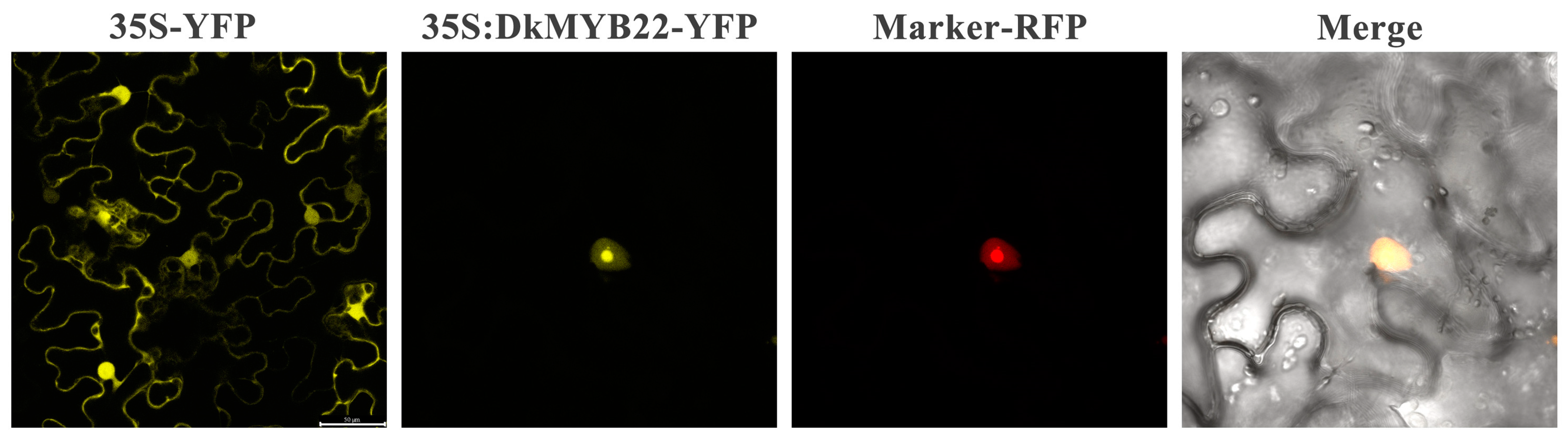
2.9. Subcellular Localization
2.10. Transient Transformation in Persimmon Leaves
3. Results
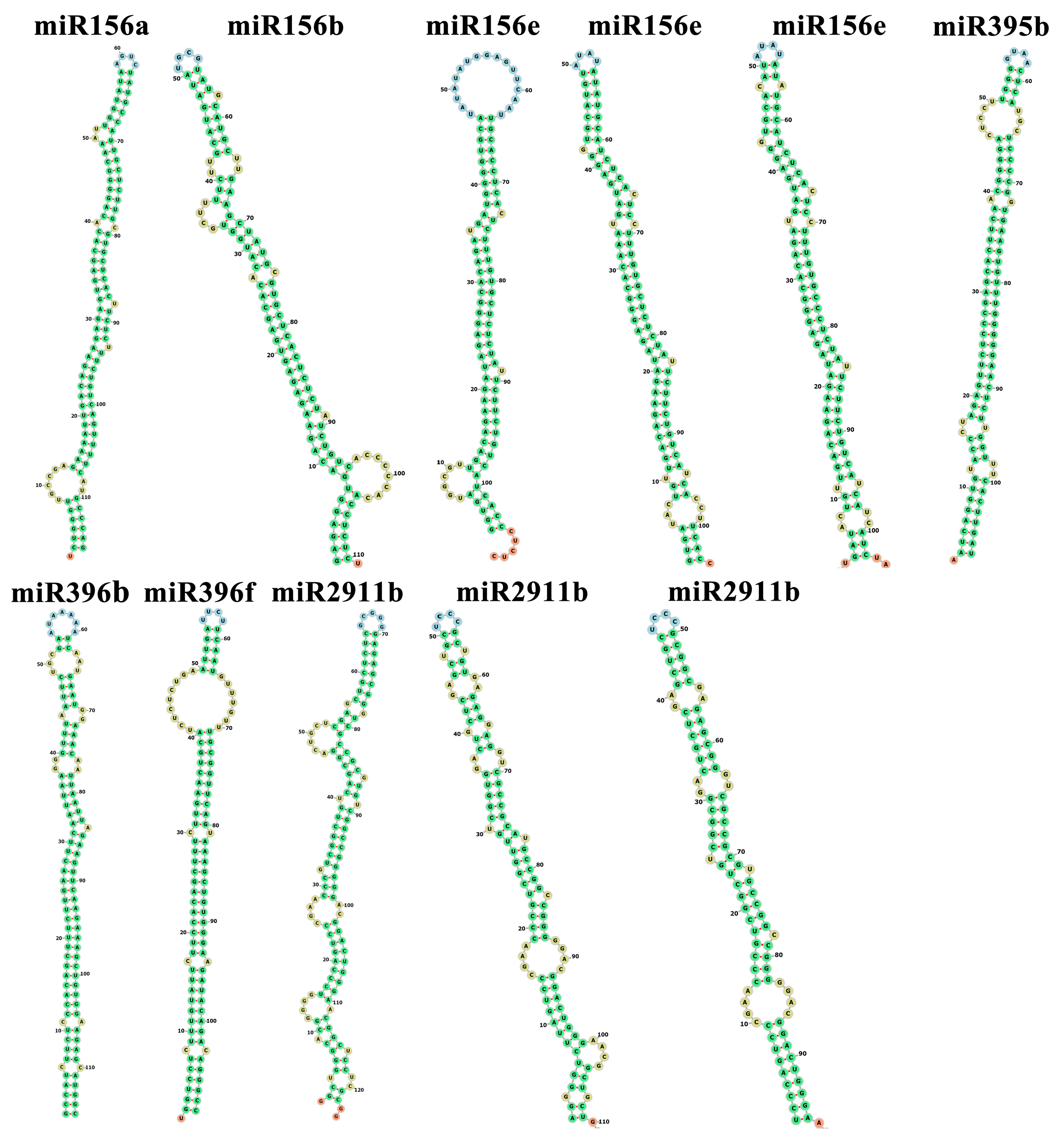
3.1. Identification of miRNA Precursors Involved in the PA Mechanism in Persimmons
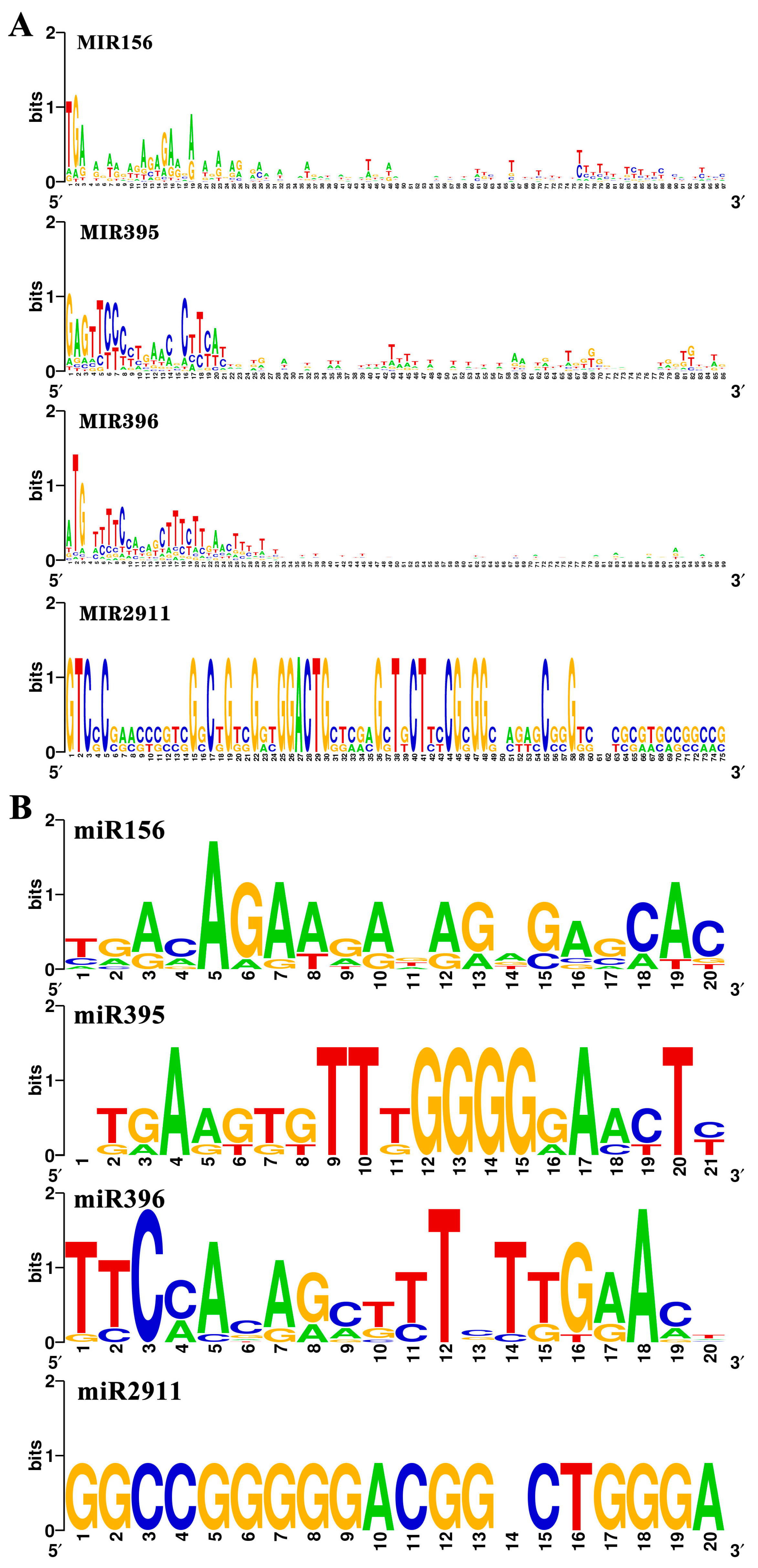
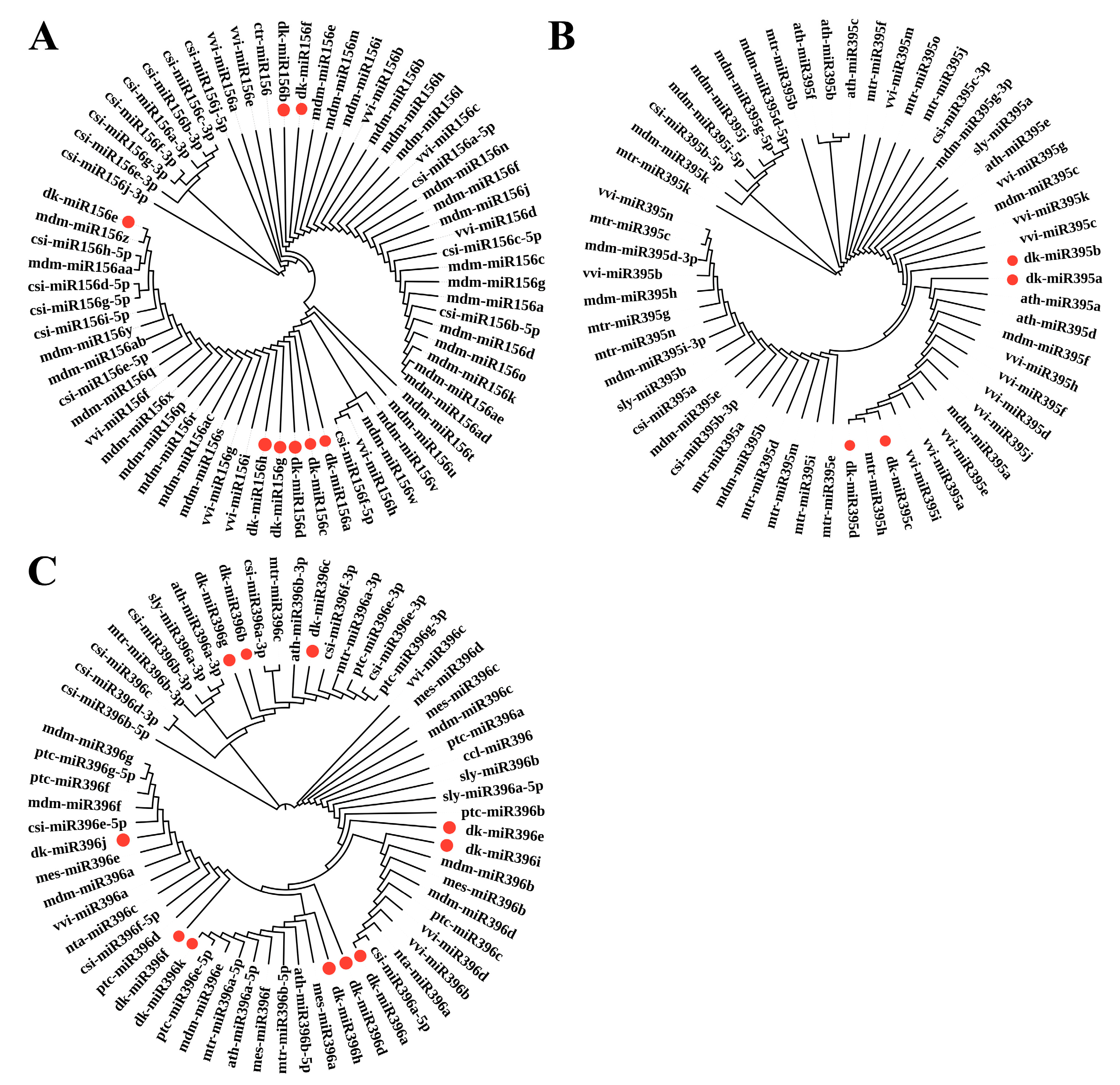
3.2. Alignments and Conservation Analysis of the miRNA Precursor Sequences in D. oleifera
3.3. MiRNA Gene Clusters in D. oleifera
3.4. MiRNA Target Prediction and Functional Analysis
3.5. Cis-Acting Elements in D. oleifera miRNA Promoters
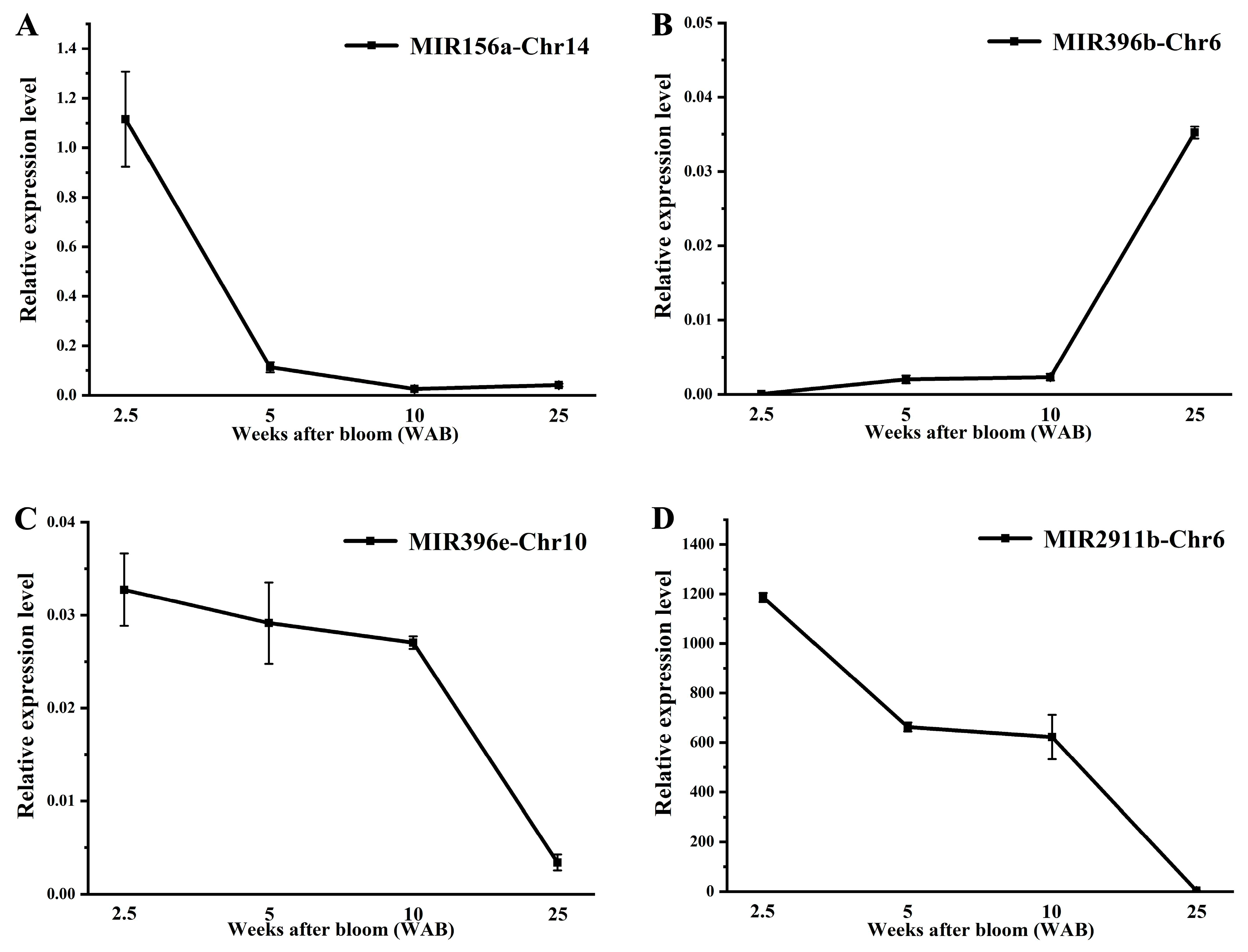
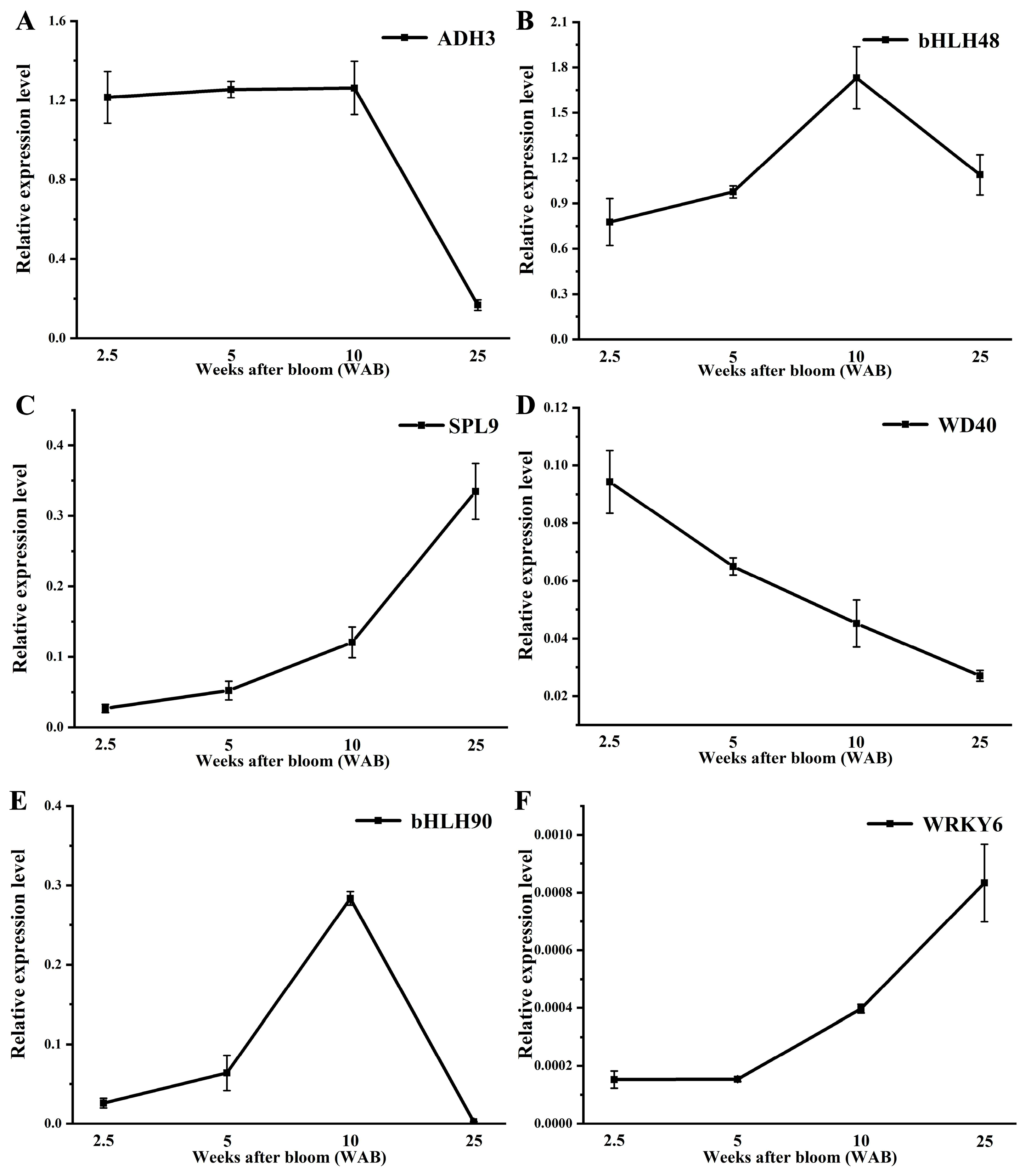
3.6. Expression Level of Pre-miRNAs and Corresponding Targets During Fruit Development in Persimmons
3.7. The Functional Analysis of the Target Gene DKMYB22 of miR2911
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Luo, Z.R.; Wang, R.Z. Persimmon in China: Domestication and traditional utilizations of genetic resources. Adv. Hort. Sci. 2008, 22, 239–243. [Google Scholar]
- Guan, C.F.; Zhang, P.X.; Hu, C.Q.; Chachar, S.; Riaz, A.; Wang, R.Z.; Yang, Y. Genetic diversity, germplasm identification and population structure of Diospyros kaki Thunb. from different geographic regions in China using SSR markers. Sci. Hortic. 2019, 251, 233–240. [Google Scholar] [CrossRef]
- Guo, D.L.; Luo, Z.R. Genetic relationships of the Japanese persimmon Diospyros kaki (Ebenaceae) and related species revealed by SSR analysis. Genet. Mol. Res. 2011, 10, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.X. Virgin avocado oil: An emerging source of functional fruit oil. J. Funct. Foods 2019, 54, 381–392. [Google Scholar] [CrossRef]
- Fu, J.M.; Liu, H.M.; Hu, J.J.; Liang, Y.Q.; Liang, J.J.; Wuyun, T.; Tan, X.F. Five complete chloroplast genome sequences from Diospyros: Genome organization and comparative analysis. PLoS ONE 2016, 11, e01595660. [Google Scholar] [CrossRef]
- Akagi, T.; Katayama-Ikegami, A.; Yonemori, K. Proanthocyanidin biosynthesis of persimmon (Diospyros Thunb.) fruit. Sci. Hortic. 2011, 130, 373–380. [Google Scholar] [CrossRef]
- Chen, W.X.; Xiong, Y.L.; Xu, L.Q.; Zhang, Q.L.; Luo, Z.R. An integrated analysis based on transcriptome and proteome reveals deastringency-related genes in CPCNA persimmon. Sci. Rep. 2017, 7, 44671. [Google Scholar] [CrossRef]
- Chen, W.X.; Zheng, Q.Y.; Li, J.W.; Liu, Y.; Xu, L.Q.; Zhang, Q.L.; Luo, Z.R. DkMYB14 is a bifunctional transcription factor that regulates the accumulation of proanthocyanidin in persimmon fruit. Plant J. 2021, 106, 1708–1727. [Google Scholar] [CrossRef]
- Mo, R.L.; Yang, S.C.; Huang, Y.M.; Chen, W.X.; Zhang, Q.L.; Luo, Z.R. ADH and PDC genes involved in tannins coagulation leading to natural de-astringency in Chinese pollination constant and non-astringency persimmon (Diospyros kaki Thunb.). Tree Genet. Genomes 2016, 12, 17. [Google Scholar] [CrossRef]
- Xu, J.C.; Ding, J.G.; Gan, J.P.; Mo, R.L.; Xu, L.Q.; Zhang, Q.L.; Luo, Z.R. ALDH2 genes are negatively correlated with natural deastringency in Chinese PCNA persimmon (Diospyros kaki Thunb.). Tree Genet. Genomes 2017, 13, 122. [Google Scholar] [CrossRef]
- Sharma, D.; Tiwari, M.; Pandey, A.; Bhatia, C.; Sharma, A.; Trivedi, P.K. MicroRNA858 is a potential regulator of phenylpropanoid pathway and plant development. Plant Physiol. 2016, 171, 944–959. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Wang, Y.Q.; Song, Z.Q.; Zhang, H.Y. Repression of MYBL2 by both microRNA858a and HY5 leads to the activa-tion of anthocyanin biosynthetic pathway in Arabidopsis. Mol. Plant 2016, 9, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Taheri-Dehkordi, A.; Naderi, R.; Martinelli, F.; Salami, S.A. Computational screening of miRNAs and their targets in saffron (Crocus sativus L.) by transcriptome mining. Planta 2021, 254, 117. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chowdhury, M.R.; Bahadur, R.P.; Basak, J. Genome-wide prediction of cauliflower miRNAs and lncRNAs and their roles in post-transcriptional gene regulation. Planta 2021, 254, 72. [Google Scholar] [CrossRef]
- Dai, X.B.; Zhuang, Z.H.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Oshida, M.; Yonemori, K.; Sugiura, A. On the nature ofcoagulated tannins in astringent-type persimmon fruit after anartificialtreatment of astringency removal. Postharvest Biol. Technol. 1996, 8, 317–327. [Google Scholar] [CrossRef]
- Mo, R.L.; Huang, Y.M.; Yang, S.C.; Zhang, Q.L.; Luo, Z.R. Development of Agrobacterium-mediated transient transformation in persimmon (Diospyros kaki Thunb.). Sci. Hortic. 2015, 192, 29–37. [Google Scholar] [CrossRef]
- Zhu, Q.G.; Xu, Y.; Yang, Y.; Guan, C.F.; Zhang, Q.Y.; Huang, J.W.; Grierson, D.; Chen, K.S.; Gong, B.C.; Yin, X.R. The persimmon (Diospyros oleifera Cheng) genome provides new insights into the inheritance of astringency and ancestral evolution. Hortic. Res. 2019, 6, 138. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Chen, W.X.; Xu, L.Q.; Guo, D.Y.; Luo, Z.R. Advance on deastringency mechanism in Chinese PCNA persimmon. Acta Hortic. 2022, 1338, 197–206. [Google Scholar] [CrossRef]
- Luo, C.; Zhang, Q.L.; Luo, Z.R. Genome-wide transcriptome analysis of Chinese pollination-constant nonastringent persimmon fruit treated with ethanol. BMC Genom. 2014, 15, 112. [Google Scholar] [CrossRef] [PubMed]
- The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.H.; Fang, Y.M.; Zhang, T.K.; Fei, Z.J.; Han, F.M.; Liu, C.Y.; Liu, M.; Xiao, W.; Zhang, W.J.; Wu, S.; et al. The pomegranate (Punica granatum L.) genome provides insights into fruit quality and ovule developmental biology. Plant Biotechnol. J. 2018, 16, 1363–1374. [Google Scholar] [CrossRef]
- Zhan, C.S.; Shen, S.Q.; Yang, C.K.; Liu, Z.H.; Fernie, A.R.; Graham, I.A.; Luo, J. Plant metabolic gene clusters in the multi-omics era. Trends Plant Sci. 2022, 27, 981–1001. [Google Scholar] [CrossRef]
- Zhang, B.H.; Pan, X.P.; Cannon, C.H.; Cobb, G.P.; Anderson, T.A. Conservation and divergence of plant microRNA genes. Plant J. 2006, 46, 243–259. [Google Scholar] [CrossRef]
- Zhu, X.D.; Leng, X.P.; Sun, X.; Mu, Q.; Wang, B.J.; Li, X.P.; Wang, C.; Fang, J.G. Discovery of conservation and diversification of miR171 genes by phylogenetic analysis based on global genomes. Plant Genome 2015, 8, 2. [Google Scholar] [CrossRef]
- Yuan, X.; Niu, J.X.; Zhou, L.; Quan, S.W.; Ma, L.; Xu, H. Evolutionary analysis of MIR156/157 family during floral induction in walnut (Juglans regia L.). J. Hortic. Sci. Biotech. 2022, 90, 697–707. [Google Scholar] [CrossRef]
- Roy, S.; Nath, D.; Paul, P.; Chakraborty, S. Computational identification of conserved microRNAs and functional annotation of their target genes in Citrus limon: Identification of microRNA in Citrus limon. S. Afr. J. Bot. 2020, 130, 109–116. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yokoyama, S.; Gato, N. Bile acid-binding activity of young persimmon (Diospyros kaki) fruit and its hypolipidemic effect in mice. Phytother. Res. 2010, 24, 205–210. [Google Scholar] [CrossRef]
- Rasmussen, S.E.; Frederiksen, H.; Krogholm, K.S.; Poulsen, L. Dietary proanthocyanidins: Occurrence, dietary intake, bioa-vailability, and protection against cardiovascular disease. Mol. Nutr. Food Res. 2005, 49, 159–174. [Google Scholar] [CrossRef]
- Gao, F.; Wang, K.; Liu, Y.; Chen, Y.P.; Chen, P.; Shi, Z.Y.; Luo, J.; Jiang, D.Q.; Fan, F.F.; Zhu, Y.G.; et al. Blocking miR396 increases rice yield by shaping inflorescence architecture. Nat. Plants 2015, 2, 15196. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.H.; Guo, S.Y.; Xu, Y.Y.; Li, C.H.; Zhang, Z.Y.; Zhang, D.J.; Xu, S.J.; Zhang, C.; Chong, K. OsmiR396d-regulated OsGRFs function in floral organogenesis in rice through binding to their targets OsJMJ706 and OsCR4. Plant Physiol. 2014, 165, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.Y.; Tan, J.; Zhou, C.; Yang, X.F.; Yang, F.; Zhang, S.J.; Sun, S.C.; Miao, X.X.; Shi, Z.Y. The OsmiR396–OsGRF8–OsF3H–flavonoid pathway mediates resistance to the brown planthopper in rice (Oryza sativa). Plant Biotechnol. J. 2019, 17, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.L.; Bian, H.W.; Zha, Y.L.; Li, F.Y.; Sun, Y.Z.; Bai, B.; Chen, Z.H.; Wang, J.H.; Zhu, M.Y.; Han, N. miR396a-mediated basic Helix–Loop–Helix transcription factor bHLH74 repression acts as a regulator for root growth in Arabidopsis seedlings. Plant Cell Physiol. 2014, 55, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Bari, A.; Orazova, S.; Ivashchenko, A. miR156- and miR171-binding sites in the protein-coding sequences of several plant genes. BioMed Res. Int. 2013, 2013, 307145. [Google Scholar] [CrossRef]
- He, J.; Xu, M.L.; Willmann, M.R.; McCormick, K.; Hu, T.Q.; Yang, L.; Starker, C.G.; Voytas, D.F.; Meyers, B.C.; Poethig, R.S. Threshold-dependent repression of SPL gene expression by miR156/miR157 controls vegetative phase change in Arabidopsis thaliana. PLoS Genet. 2018, 14, e1007337. [Google Scholar] [CrossRef]
- Wu, X.M.; Liu, M.Y.; Xu, Q.; Guo, W.W. Identification and characterization of micro RNAs from citrus expressed sequence tags. Tree Genet. Genomes 2011, 7, 117–133. [Google Scholar] [CrossRef]
- Gou, J.Y.; Felippes, F.F.; Liu, C.J.; Weigel, D.; Wang, J.W. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 2011, 23, 1512–1522. [Google Scholar] [CrossRef]
- Guan, C.F.; Wang, M.K.; Zhang, Y.F.; Ruan, X.F.; Zhang, Q.L.; Luo, Z.R.; Yang, Y. DkWRKY interacts with pyruvate kinase gene DkPK1 and promotes natural deastringency in C-PCNA persimmon. Plant Sci. 2020, 290, 110285. [Google Scholar] [CrossRef]
- Zhou, M.; Sun, J.; Wang, Q.H.; Song, L.Q.; Zhao, G.; Wang, H.Z.; Yang, H.X.; Li, X. Genome-wide analysis of clustering patterns and flanking characteristics for plant microRNA genes. FEBS J. 2011, 278, 929–940. [Google Scholar] [CrossRef]
- Chen, M.; Wei, M.; Dong, Z.H.; Bao, H.; Wang, Y.W. Genomic identification of microRNA promoters and their cis-acting elements in Populus. Genes Genom. 2016, 38, 377–387. [Google Scholar] [CrossRef]
- Barvkar, V.T.; Pardeshi, V.C.; Kale, S.M.; Qiu, S.Q.; Rollins, M.; Datla, R.; Gupta, V.S.; Kadoo, N.Y. Genome-wide identifica-tion and characterization of microRNA genes and their targets in flax (Linum usitatissimum). Planta 2013, 237, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, Y.X.; Han, Y.P.; Li, Y.G.; Guo, M.Z.; Li, W.B. MicroRNA primary transcripts and promoter elements analysis in soybean (Glycine max L. Merril.). J. Integr. Agric. 2013, 12, 1522–1529. [Google Scholar] [CrossRef]
- Kong, W.W.; Yang, Z.M. Identification of iron-deficiency responsive microRNA genes and cis-elements in Arabidopsis. Plant Physiol. Biochem. 2010, 48, 153–159. [Google Scholar] [CrossRef]
- Zhao, X.; Li, L. Comparative analysis of microRNA promoters in Arabidopsis and rice. Genom. Proteom. Bioinform. 2013, 11, 56–60. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNAs | Target Transcripts | Description |
|---|---|---|
| miR156 | EVM0020578.2 | Squamosa promoter-binding-like protein 18 |
| EVM0013648.1 | ||
| EVM0005209.1 | Transcription factor MYB4 | |
| EVM0014735.1 | Alcohol dehydrogenase 3 | |
| EVM0025134.1 | ||
| EVM0016011.1 | ||
| EVM0008150.1 | ||
| EVM0009573.1 | ||
| EVM0031798.1 | F-box-like/WD repeat-containing protein | |
| EVM0024972.1 | Squamosa promoter-binding-like protein 13A | |
| EVM0006611.3 | Squamosa promoter-binding-like protein 16 | |
| EVM0031300.1 | Squamosa promoter-binding-like protein 9 | |
| EVM0022316.1 | ||
| EVM0012089.2 | Squamosa promoter-binding-like protein 2 | |
| EVM0031388.1 | ||
| EVM0021122.1 | Squamosa promoter-binding-like protein 6 | |
| EVM0016005.3 | ||
| EVM0014405.1 | Squamosa promoter-binding-like protein 7 | |
| EVM0025944.2 | Basic helix-loop-helix DNA-binding superfamily protein isoform 1 | |
| EVM0012958.1 | Transcription factor bHLH48 | |
| miR395 | EVM0006832.1 | Transcription factor bHLH147 |
| miR396 | EVM0004765.1 | Transcription factor bHLH90 |
| EVM0013146.1 | WD repeat-containing protein 74 | |
| EVM0022732.1 | Diospyros kaki pyruvate decarboxylase 2 (PDC2) mRNA | |
| EVM0031521.1 | Laccase-11-like | |
| EVM0031715.1 | Diospyros kaki ethylene response factor 3 (ERF3) mRNA | |
| EVM0011729.1 | WD repeat-containing protein 75 | |
| EVM0021485.1 | F-box/WD-40 repeat-containing protein | |
| EVM0019662.1 | Transmembrane 9 superfamily member 1 | |
| miR2911 | EVM0018100.1 | Ethylene-responsive transcription factor TINY-like |
| EVM0030558.1 | Ethylene-responsive transcription factor CRF2 | |
| EVM0005439.1 | Transcription factor MYB22 | |
| EVM0014721.1 | WD repeat-containing protein 44-like | |
| EVM0019487.1 | Probable WRKY transcription factor 72 | |
| EVM0030913.1 | WRKY transcription factor 6 partial mRNA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Wu, R.; Hu, X.; Luo, Z.; Zhang, Q.; Yang, S. Genome-Wide Identification of miRNAs in Oily Persimmon (Diospyros oleifera Cheng) and Their Functional Targets Associated with Proanthocyanidin Metabolism. Horticulturae 2025, 11, 41. https://doi.org/10.3390/horticulturae11010041
Zhang M, Wu R, Hu X, Luo Z, Zhang Q, Yang S. Genome-Wide Identification of miRNAs in Oily Persimmon (Diospyros oleifera Cheng) and Their Functional Targets Associated with Proanthocyanidin Metabolism. Horticulturae. 2025; 11(1):41. https://doi.org/10.3390/horticulturae11010041
Chicago/Turabian StyleZhang, Meng, Rong Wu, Xinlong Hu, Zhengrong Luo, Qinglin Zhang, and Sichao Yang. 2025. "Genome-Wide Identification of miRNAs in Oily Persimmon (Diospyros oleifera Cheng) and Their Functional Targets Associated with Proanthocyanidin Metabolism" Horticulturae 11, no. 1: 41. https://doi.org/10.3390/horticulturae11010041
APA StyleZhang, M., Wu, R., Hu, X., Luo, Z., Zhang, Q., & Yang, S. (2025). Genome-Wide Identification of miRNAs in Oily Persimmon (Diospyros oleifera Cheng) and Their Functional Targets Associated with Proanthocyanidin Metabolism. Horticulturae, 11(1), 41. https://doi.org/10.3390/horticulturae11010041