
Comparative Transcriptomic Analysis of Pyrethrin and EβF Biosynthesis in Tanacetum cinerariifolium Stems and Flowers



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
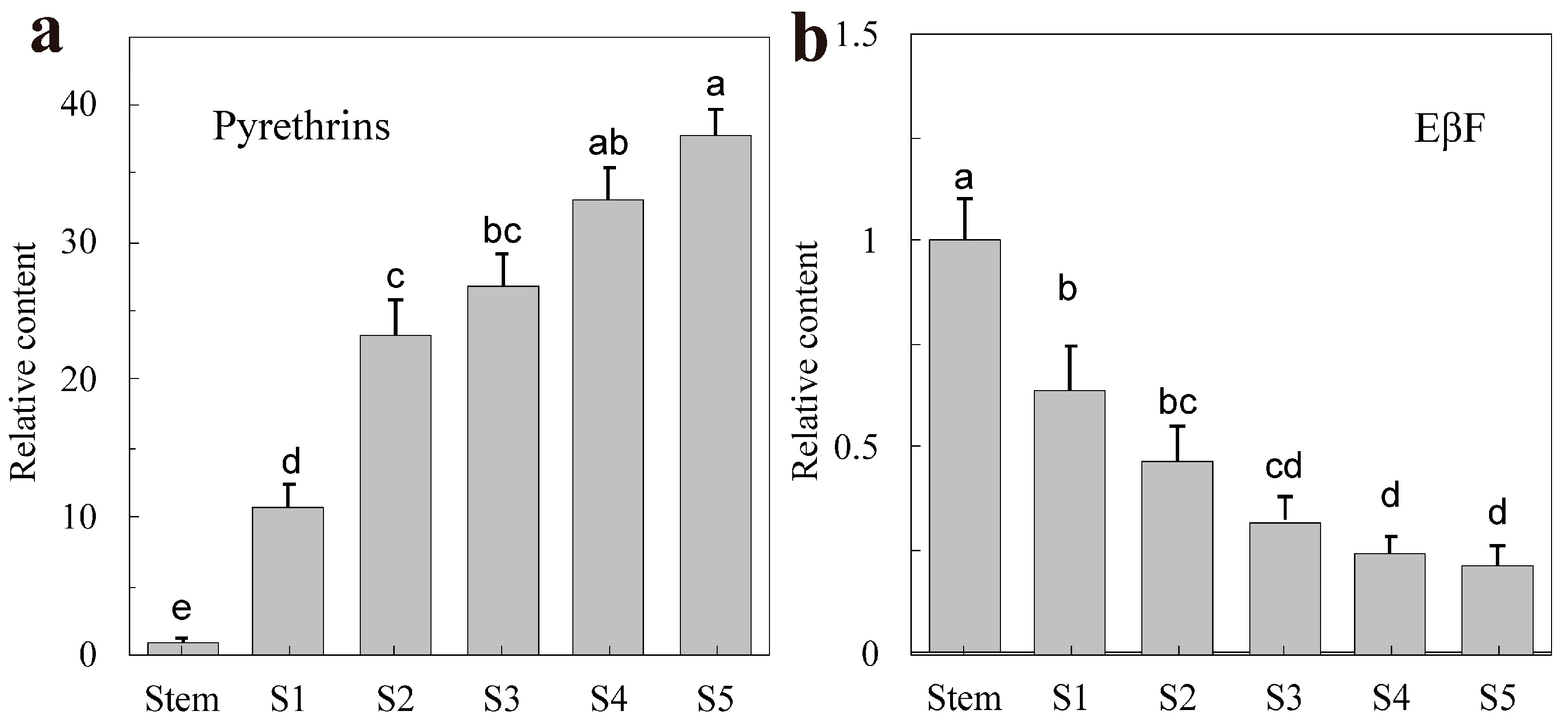
2.1. The Content of Pyrethrin and EβF in T. cinerariifolium
2.2. Transcriptome Sequencing, Assembly, and Annotation of T. cinerariifolium
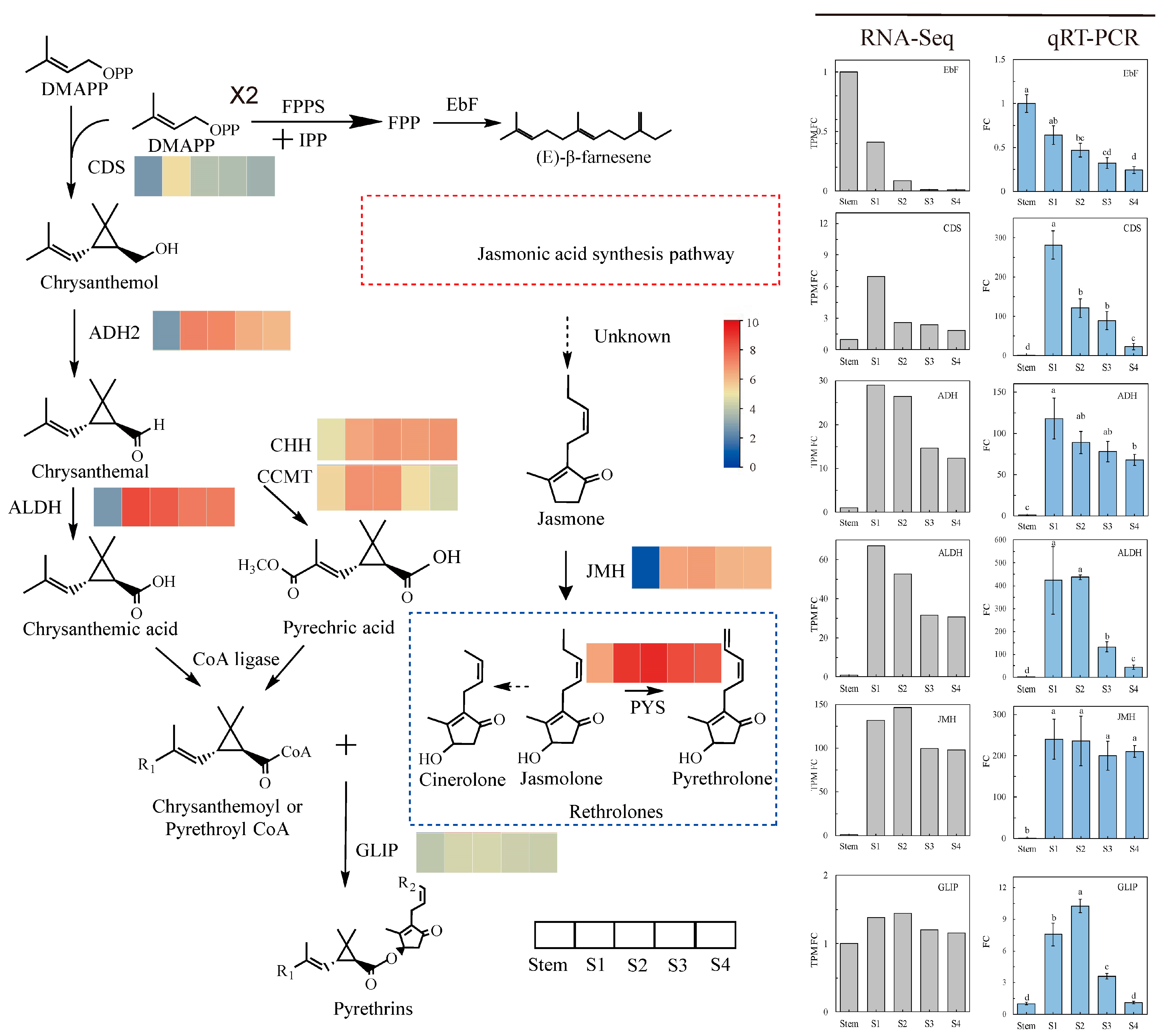
2.3. Differential Gene Expression in JA Biosynthesis
2.4. Differential Gene Expression in Pyrethrins Biosynthesis
2.5. WGCNA Co-Expression Analysis
3. Discussion
3.1. Dynamics of Pyrethrin and EβF Biosynthesis in T. cinerariifolium
3.2. Insights from Co-Expression Analysis
3.3. Implications for Pest Management and Future Directions
4. Materials and Methods
4.1. Plant Materials
4.2. GC-MS Analysis
4.3. Total RNA Extraction, cDNA Library Construction and Sequencing
4.4. Transcript Splicing, Annotation, and Quality Assessment
4.5. Screening and Enrichment Analysis of Differential Genes
4.6. WGNCA Analysis
4.7. Transcriptome qRT-PCR Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mossa, A.H.; Mohafrash, S.M.M.; Chandrasekaran, N. Safety of natural insecticides: Toxic effects on experimental animals. BioMed Res. Int. 2018, 2018, 4308054. [Google Scholar] [CrossRef] [PubMed]
- Lybrand, D.B.; Xu, H.; Last, R.L.; Pichersky, E. How plants synthesize pyrethrins: Safe and biodegradable insecticides. Trends Plant Sci. 2020, 25, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K. Chemical and biological studies of natural and synthetic products for the highly selective control of pest insect species. Biosci. Biotechnol. Biochem. 2022, 86, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Grdisa, M.; Carovic-Stanko, K.; Kolak, I.; Satovic, Z. Morphological and biochemical diversity of dalmatian pyrethrum (Tanacetum cinerariifolium (Trevir.) Sch Bip.). Agric. Conspec. Sci. 2009, 74, 73–80. [Google Scholar]
- Jeran, N.; Grdiša, M.; Varga, F.; Šatović, Z.; Liber, Z.; Dabić, D.; Biošić, M. Pyrethrin from Dalmatian pyrethrum (Tanacetum cinerariifolium/Trevir./Sch. Bip.): Biosynthesis, biological activity, methods of extraction and determination. Phytochem. Rev. 2021, 20, 875–905. [Google Scholar] [CrossRef]
- Zeng, T.; Li, J.; Li, J.; Hu, H.; Zhu, L.; Liu, K.; Bai, J.; Jiang, Q.; Wang, C. Pyrethrins in Tanacetum cinerariifolium: Biosynthesis, regulation, and agricultural application. Ornam. Plant Res. 2024, 4, e015. [Google Scholar] [CrossRef]
- Li, J.J.; Hu, H.; Chen, Y.; Xie, J.; Li, J.; Zeng, T.; Wang, M.Q.; Luo, J.; Zheng, R.R.; Jongsma, M.A.; et al. Tissue specificity of (E)-beta-farnesene and germacrene D accumulation in pyrethrum flowers. Phytochemistry 2021, 187, 112768. [Google Scholar] [CrossRef]
- Li, J.J.; Hu, H.; Mao, J.; Yu, L.; Stoopen, G.; Wang, M.Q.; Mumm, R.; de Ruijter, N.C.A.; Dicke, M.; Jongsma, M.A.; et al. Defense of pyrethrum flowers: Repelling herbivores and recruiting carnivores by producing aphid alarm pheromone. New Phytol. 2019, 223, 1607–1620. [Google Scholar] [CrossRef] [PubMed]
- Vandermoten, S.; Mescher, M.C.; Francis, F.; Haubruge, E.; Verheggen, F.J. Aphid alarm pheromone: An overview of current knowledge on biosynthesis and functions. Insect Biochem. Mol. Biol. 2012, 42, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, B.; Grossi, G.; Falabella, P.; Liu, Y.; Yan, S.; Lu, J.; Xi, J.; Wang, G. Molecular basis of alarm pheromone detection in aphids. Curr. Biol. 2017, 27, 55–61. [Google Scholar] [CrossRef]
- Li, J.; Hu, H.; Ren, S.; Yu, L.; Luo, Y.; Li, J.; Zeng, T.; Wang, M.; Wang, C. Aphid alarm pheromone mimicry in transgenic Chrysanthemum morifolium: Insights into the potential of (E)-β-farnesene for aphid resistance. Front. Plant Sci. 2024, 15, 1373669. [Google Scholar] [CrossRef] [PubMed]
- Bhat, B.K.; Menary, R.C. Scanning electron microscopic study of oil glands in pyrethrum flowers. Pyrethrum Post 1975, 15, 11–15. [Google Scholar]
- Head, S.W. A study of the insecticidal constituents in Chrysanthemum cinerariaefolium. (1) Their development in the flower head. (2) Their distribution in the plant. Pyrethrum Post 1966, 8, 32–37. [Google Scholar]
- Kikuta, Y.; Ueda, H.; Nakayama, K.; Katsuda, Y.; Ozawa, R.; Takabayashi, J.; Hatanaka, A.; Matsuda, K. Specific regulation of pyrethrin biosynthesis in Chrysanthemum cinerariaefolium by a blend of volatiles emitted from artificially damaged conspecific plants. Plant Cell Physiol. 2011, 52, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Kikuta, Y.; Matsuda, K. Plant communication: Mediated by individual or blended VOCs? Plant Signal. Behav. 2012, 7, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Z.; Cui, Z.; Qi, Q.; Hou, J. Progress and perspectives for microbial production of farnesene. Bioresour. Technol. 2022, 347, 126682. [Google Scholar] [CrossRef]
- Matsuda, K.; Kikuta, Y.; Haba, A.; Nakayama, K.; Katsuda, Y.; Hatanaka, A.; Komai, K. Biosynthesis of pyrethrin I in seedlings of Chrysanthemum cinerariaefolium. Phytochemistry 2005, 66, 1529–1535. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Liu, F.; Wang, Q.; Xu, P.; Andreazza, F.; Valbon, W.R.; Bandason, E.; Chen, M.; Yan, R.; Feng, B.; Smith, L.B.; et al. A dual-target molecular mechanism of pyrethrum repellency against mosquitoes. Nat. Commun. 2021, 12, 2553. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Li, J.W.; Xu, Z.Z.; Zhou, L.; Li, J.J.; Yu, Q.; Luo, J.; Chan, Z.L.; Jongsma, M.A.; Hu, H.; et al. TcMYC2 regulates pyrethrin biosynthesis in Tanacetum cinerariifolium. Hortic. Res. 2022, 9, uhac178. [Google Scholar] [CrossRef]
- Zito, S.W.; Zieg, R.G.; Staba, E.J. Distribution of pyrethrins in oil glands and leaf tissue of Chrysanthemum cinerariaefolium. Planta Medica 1983, 47, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; He, Z.J.; He, J.F.; Lv, W.; Huang, S.X.; Li, J.W.; Zhu, L.Y.; Wan, S.; Zhou, W.F.; Yang, Z.S.; et al. The Telomere-to-telomere gap-free reference genome of wild blueberry (Vaccinium duclouxii) provides its high soluble sugar and anthocyanin accumulation. Hortic. Res. 2023, 10, uhad209. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wu, Y.; Gao, R.; Zhang, Y.; Zheng, R.; Fang, M.; Li, Y.; Zhang, Y.; Guan, L.; Gao, Y. Integrated metabolomics and transcriptomics reveal the key role of flavonoids in the cold tolerance of chrysanthemum. Int. J. Mol. Sci. 2024, 25, 7589. [Google Scholar] [CrossRef] [PubMed]
- Ting, Z.; Pengcheng, Z.; Jiang, X.; Lingzhu, W.; Wanting, S.; Jiang, W.; Jianhui, C. Integrated transcriptomic and metabolomic analysis reveals the effects of forchlorfenuron and thidiazuron on flavonoid biosynthesis in table grape skins. Curr. Plant Biol. 2024, 40, 100417. [Google Scholar]
- Li, J.J.; Hu, H.; Fu, H.S.; Li, J.; Zeng, T.; Li, J.W.; Wang, M.Q.; Jongsma, M.A.; Wang, C.Y. Exploring the co-operativity of secretory structures for defense and pollination in flowering plants. Planta 2024, 259, 41. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Luo, Y.; Li, M.; Li, J.; Zeng, T.; Luo, J.; Chang, X.; Wang, M.; Jongsma, M.A.; Hu, H.; et al. Nocturnal burst emissions of germacrene D from the open disk florets of pyrethrum flowers induce moths to oviposit on a nonhost and improve pollination success. New Phytol. 2024, 244, 2036–2048. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, D.; Groom, T.; Nicolas, M. Pattern of pyrethrin accumulation, achene and trichome development in relation to pattern of flower development in pyrethrum. II Int. Symp. Pyrethrum 2015, 1169, 93–100. [Google Scholar] [CrossRef]
- Li, J.W.; Zeng, T.; Xu, Z.Z.; Li, J.J.; Hu, H.; Yu, Q.; Zhou, L.; Zheng, R.R.; Luo, J.; Wang, C.Y. Ribozyme-mediated CRISPR/Cas9 gene editing in pyrethrum (Tanacetum cinerariifolium) hairy roots using a RNA polymerase II-dependent promoter. Plant Methods 2022, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Wang, S. TRANSPARENT TESTA GLABRA1, a key regulator in plants with multiple roles and multiple function mechanisms. Int. J. Mol. Sci. 2020, 21, 4881. [Google Scholar] [CrossRef] [PubMed]
- Pesch, M.; Dartan, B.; Birkenbihl, R.; Somssich, I.E.; Hülskamp, M. Arabidopsis TTG2 regulates TRY expression through enhancement of activator complex-triggered activation. Plant Cell 2014, 26, 4067–4083. [Google Scholar] [CrossRef]
- Zhao, M.; Morohashi, K.; Hatlestad, G.; Grotewold, E.; Lloyd, A. The TTG1-bHLH-MYB complex controls trichome cell fate and patterning through direct targeting of regulatory loci. Development 2008, 135, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.H.; Yan, T.X.; Li, L.; Chen, M.H.; Ma, Y.N.; Hao, X.L.; Fu, X.Q.; Shen, Q.; Huang, Y.W.; Qin, W.; et al. The WRKY transcription factor AaGSW2 promotes glandular trichome initiation in Artemisia annua. J. Exp. Bot. 2020, 72, 1691–1701. [Google Scholar] [CrossRef]
- Hu, D.G.; Li, Y.Y.; Zhang, Q.Y.; Li, M.; Sun, C.H.; Yu, J.Q.; Hao, Y.J. The R2R3-MYB transcription factor MdMYB73 is involved in malate accumulation and vacuolar acidification in apple. Plant J. 2017, 91, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.M. Pyrethrum Secondary Metabolism: Biosynthesis, Localization and Ecology of Defence Compounds. Ph.D. Thesis, Wageningen University, Wageningen, The Netherlands, 2013. [Google Scholar]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 19, 3210–3212. [Google Scholar] [CrossRef]
- Tian, F.; Yang, D.C.; Meng, Y.Q.; Jin, J.P.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, H.; Liu, Y.; Liu, Y.X.; Huang, L. EVenn: Easy to create repeatable and editable Venn diagrams and Venn networks online. J. Genet. Genom. 2021, 48, 863–866. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 16. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. CytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 408, 402–408. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, T.; Li, J.; Wang, C.; Li, J. Comparative Transcriptomic Analysis of Pyrethrin and EβF Biosynthesis in Tanacetum cinerariifolium Stems and Flowers. Horticulturae 2025, 11, 201. https://doi.org/10.3390/horticulturae11020201
Zeng T, Li J, Wang C, Li J. Comparative Transcriptomic Analysis of Pyrethrin and EβF Biosynthesis in Tanacetum cinerariifolium Stems and Flowers. Horticulturae. 2025; 11(2):201. https://doi.org/10.3390/horticulturae11020201
Chicago/Turabian StyleZeng, Tuo, Jiawen Li, Caiyun Wang, and Jinjin Li. 2025. "Comparative Transcriptomic Analysis of Pyrethrin and EβF Biosynthesis in Tanacetum cinerariifolium Stems and Flowers" Horticulturae 11, no. 2: 201. https://doi.org/10.3390/horticulturae11020201
APA StyleZeng, T., Li, J., Wang, C., & Li, J. (2025). Comparative Transcriptomic Analysis of Pyrethrin and EβF Biosynthesis in Tanacetum cinerariifolium Stems and Flowers. Horticulturae, 11(2), 201. https://doi.org/10.3390/horticulturae11020201