
Genomic and Transcriptomic Analysis to Explore the Biological Characteristics of Cyclocybe chaxingu
 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Species Information
2.2. Strains and Culture Conditions of Cyclocybe chaxingu
2.3. Genome Sequencing and Assembly
2.4. Gene Prediction and Annotation
2.5. Comparative Genomic Analysis
2.6. Analysis of Mating Genes
2.7. Transcriptomic Analysis of Different Tissues of Cyclocybe chaxingu
3. Results and Discussion
3.1. Cultivation of Cyclocybe chaxingu and Monokaryotic Strain Isolation
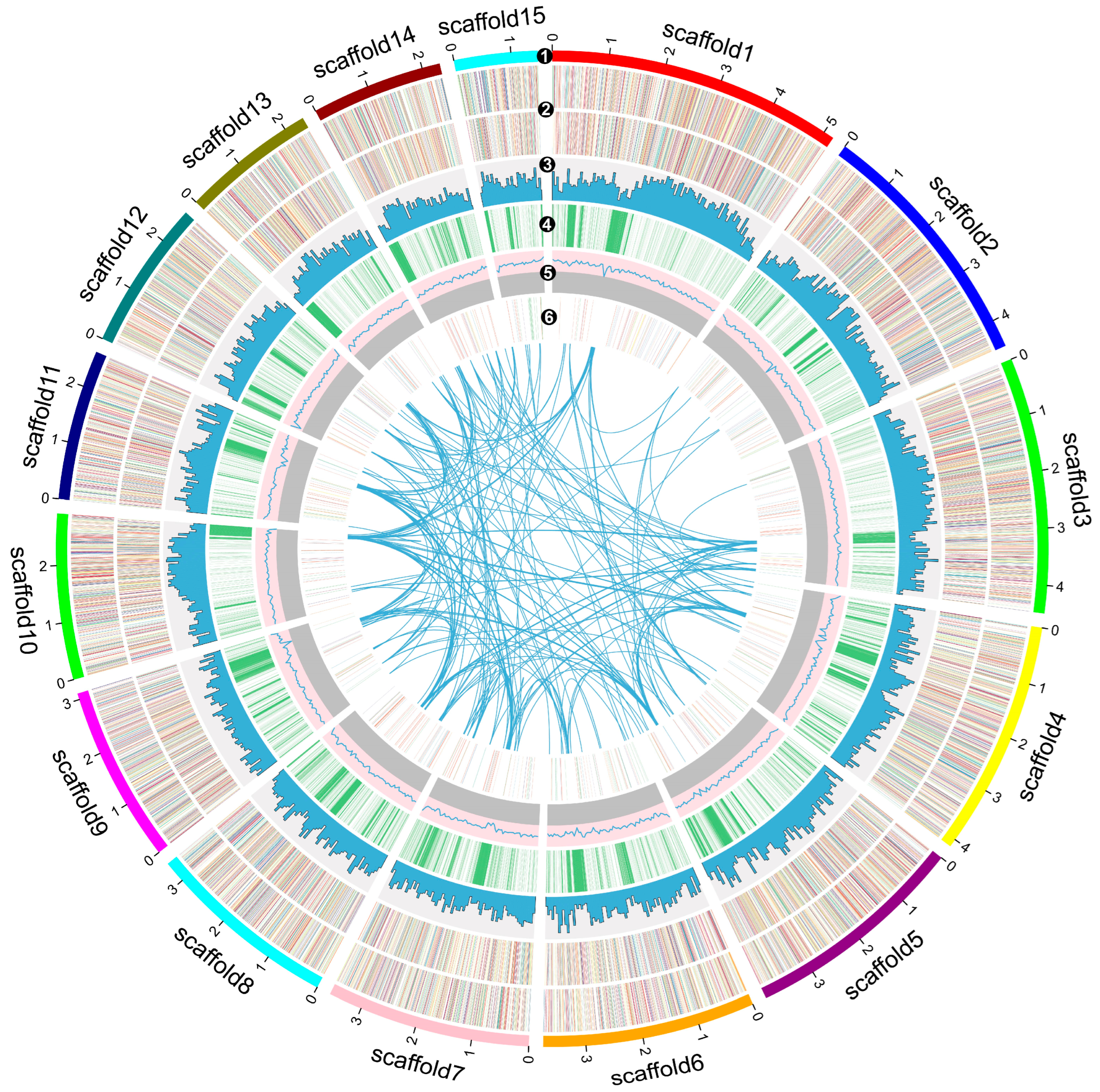
3.2. Genome Assembly, Prediction, and Annotation of Cyclocybe chaxingu
3.3. Genome Evolutionary and Comparative Analysis
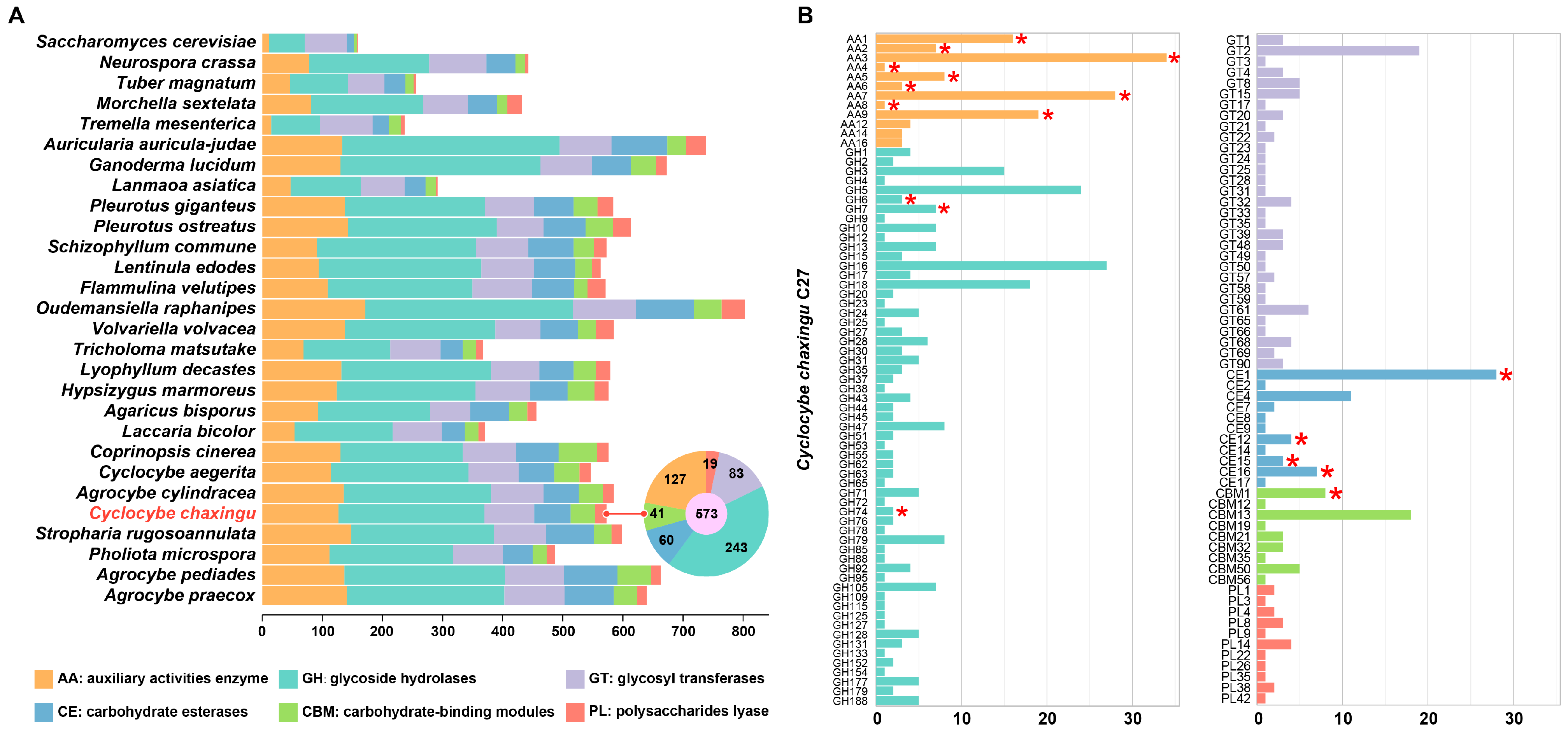
3.4. Carbohydrate Active Enzymes in Cyclocybe chaxingu Genome
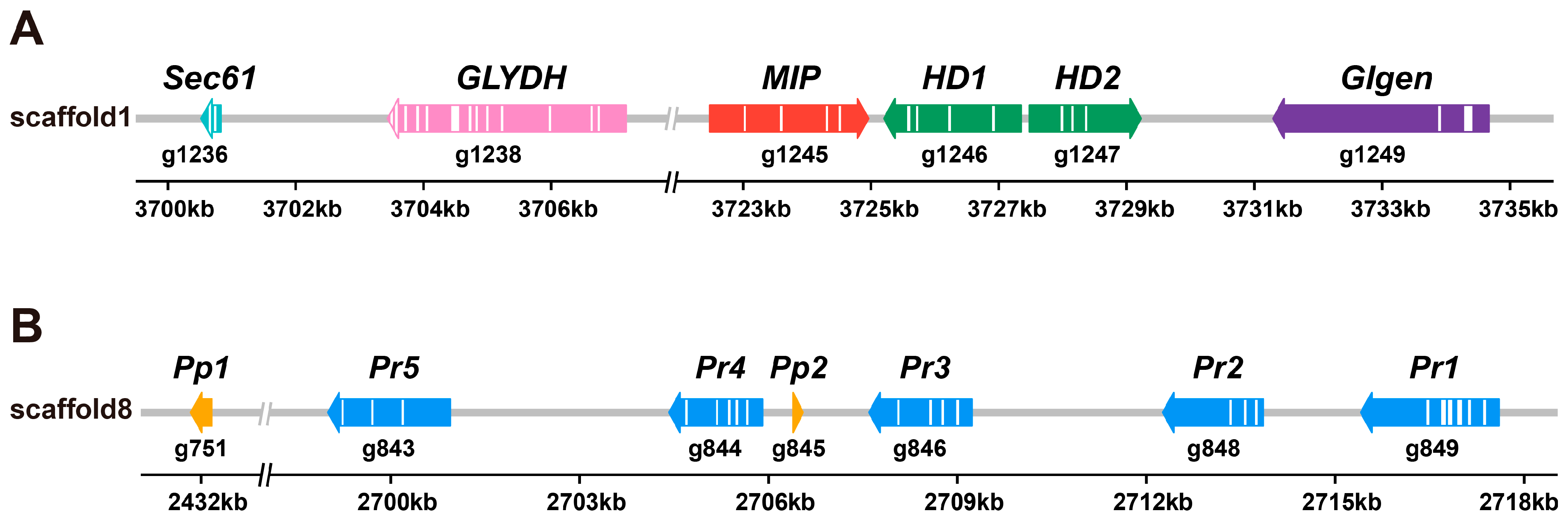
3.5. Identification of the Mating Locus
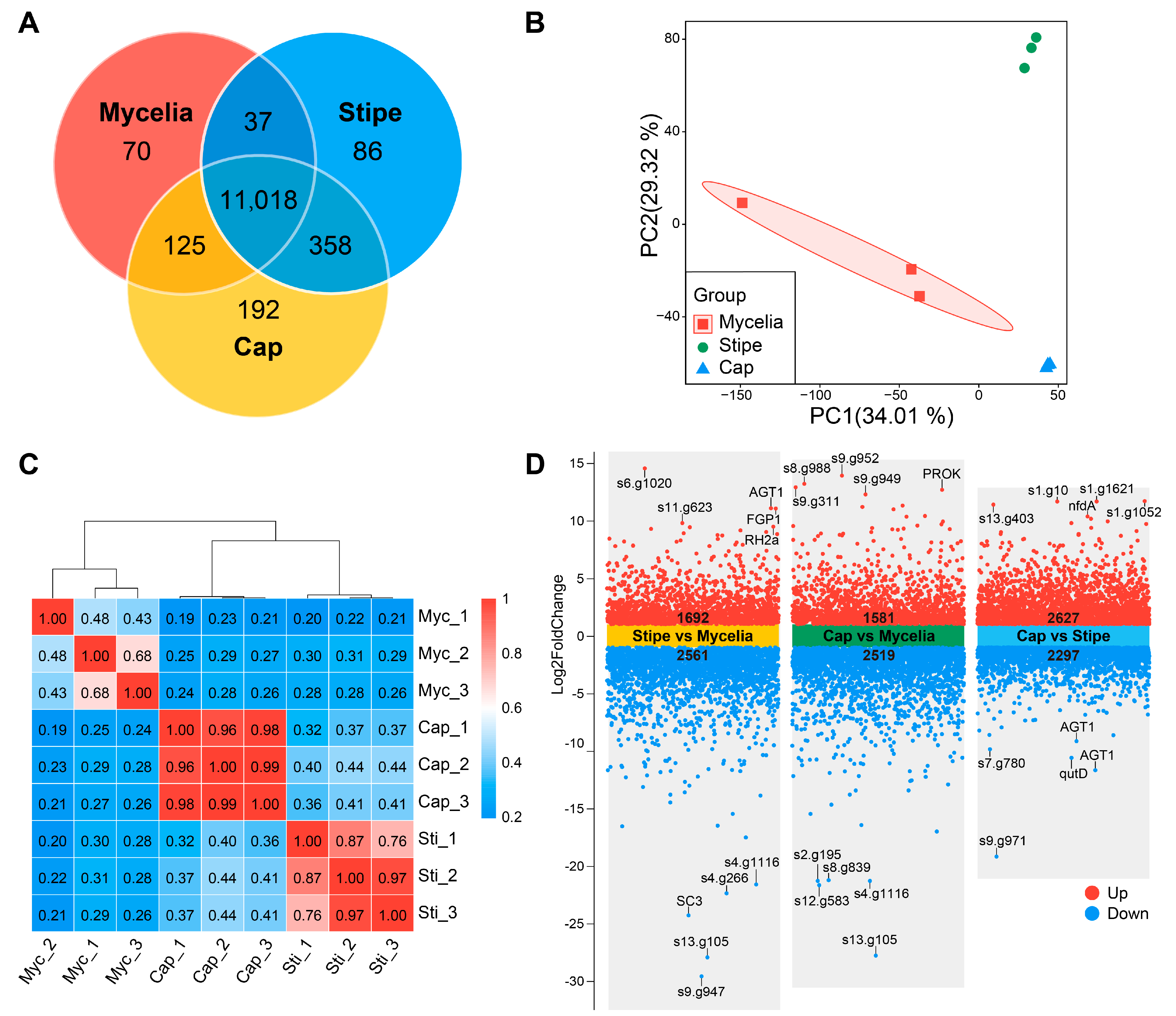
3.6. Transcriptome Data Analysis
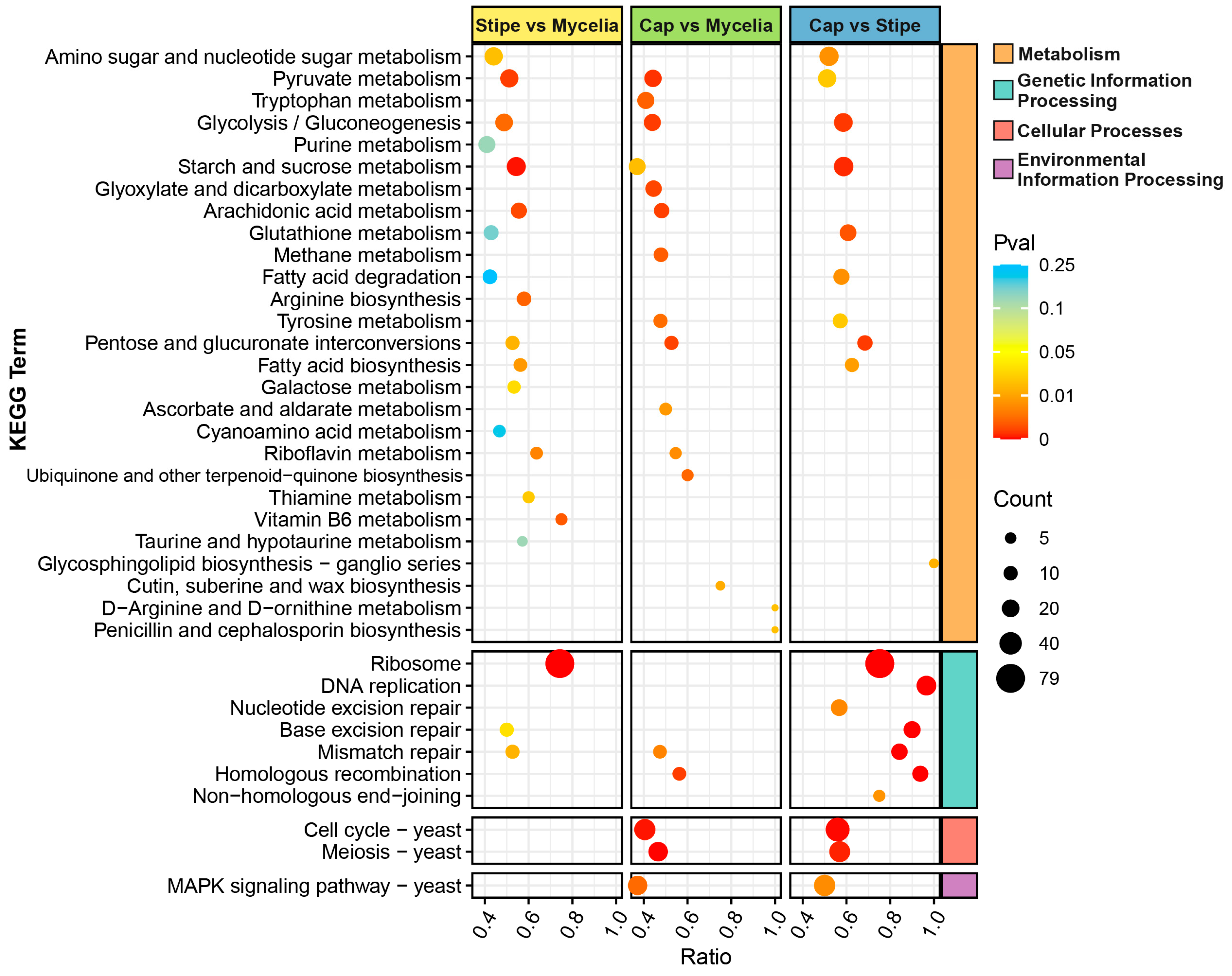
3.7. Differential Expression and Gene Enrichment Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, Q.; Song, H.; Chen, R.; Chen, M.; Zhai, Z.; Zhou, J.; Gao, Y.; Hu, D. Species concept of Cyclocybe chaxingu, an edible mushroom cultivated in China. Mycosystema 2021, 40, 981–991. [Google Scholar] [CrossRef]
- Liu, Q.; Hua, R.; Zhang, J.; Li, J.; Shang, L.; Luo, X.; Liu, S. Biological characteristics, genetic breeding and cultivation situation of Cyclocybe chaxingu. Edible Med. Mushrooms 2021, 29, 501–505. [Google Scholar]
- Liu, Q.; Liu, Y.; Chen, M.; Zhai, Z.; Zhou, J.; Hu, D. Study on the Systematics of the Genus Agrocybe. Edible Fungi China 2020, 39, 1–7+16. [Google Scholar] [CrossRef]
- Xu, J.; Wang, S.; Gong, X.; Yu, R.; Wang, L. Composition analysis of Agrocybe cylindracea extract. Sci. Technol. Food Ind. 2006, 27, 165–167. [Google Scholar] [CrossRef]
- Lee, K.J.; Yun, I.J.; Kim, K.H.; Lim, S.H.; Ham, H.J.; Eum, W.S.; Joo, J.H. Amino acid and fatty acid compositions of Agrocybe chaxingu, an edible mushroom. J. Food Compos. Anal. 2011, 24, 175–178. [Google Scholar] [CrossRef]
- Zhao, H.; Li, J.; Zhang, J.; Wang, X.; Hao, L.; Jia, L. Purification, in vitro antioxidant and in vivo anti-aging activities of exopolysaccharides by Agrocybe cylindracea. Int. J. Biol. Macromol. 2017, 102, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Kiho, T.; Yoshida, I.; Nagai, K.; Ukai, S.; Hara, C. (1→3)-α-d-glucan from an alkaline extract of Agrocybe cylindracea, and antitumor activity of its O-(carboxymethyl)ated derivatives. Carbohydr. Res. 1989, 189, 273–279. [Google Scholar] [CrossRef]
- Morin, E.; Kohler, A.; Baker, A.R.; Foulongne-Oriol, M.; Lombard, V.; Nagy, L.G.; Ohm, R.A.; Patyshakuliyeva, A.; Brun, A.; Aerts, A.L.; et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci. USA 2012, 109, 17501–17506. [Google Scholar] [CrossRef]
- Martin, F.; Aerts, A.; Ahren, D.; Brun, A.; Danchin, E.G.J.; Duchaussoy, F.; Gibon, J.; Kohler, A.; Lindquist, E.; Pereda, V.; et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 2008, 452, 88–92. [Google Scholar] [CrossRef]
- Stajich, J.E.; Wilke, S.K.; Ahrén, D.; Au, C.H.; Birren, B.W.; Borodovsky, M.; Burns, C.; Canbäck, B.; Casselton, L.A.; Cheng, C.K.; et al. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc. Natl. Acad. Sci. USA 2010, 107, 11889–11894. [Google Scholar] [CrossRef]
- Ohm, R.A.; de Jong, J.F.; Lugones, L.G.; Aerts, A.; Kothe, E.; Stajich, J.E.; de Vries, R.P.; Record, E.; Levasseur, A.; Baker, S.E.; et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 2010, 28, 957–963. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, M.; Sun, Y.; Li, Q.; Liu, J.; Song, C.; Shang, X.; Tan, Q.; Zhang, L.; Yu, H. Whole-genome sequence of a high-temperature edible mushroom Pleurotus giganteus (zhudugu). Front. Microbiol. 2022, 13, 941889. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Baek, J.H.; Lee, S.; Kim, C.; Rhee, H.; Kim, H.; Seo, J.; Park, H.; Yoon, D.; Nam, J.; et al. Whole genome and global gene expression analyses of the model mushroom Flammulina velutipes reveal a high capacity for lignocellulose degradation. PLoS ONE 2014, 9, e93560. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gong, Y.; Cai, Y.; Liu, W.; Zhou, Y.; Xiao, Y.; Xu, Z.; Liu, Y.; Lei, X.; Wang, G.; et al. Genome Sequence of the Edible Cultivated Mushroom Lentinula edodes (Shiitake) Reveals Insights into Lignocellulose Degradation. PLoS ONE 2016, 11, e0160336. [Google Scholar] [CrossRef]
- Zhu, L.; Gao, X.; Zhang, M.; Hu, C.; Yang, W.; Guo, L.; Yang, S.; Yu, H.; Yu, H. Whole Genome Sequence of an Edible Mushroom Oudemansiella raphanipes (Changgengu). J. Fungi 2023, 9, 266. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, W.; Qiu, T.; Gao, X.; Zhang, H.; Zhang, S.; Cui, H.; Guo, L.; Yu, H.; Yu, H. Complete genome sequences and comparative secretomic analysis for the industrially cultivated edible mushroom Lyophyllum decastes reveals insights on evolution and lignocellulose degradation potential. Front. Microbiol. 2023, 14, 1137162. [Google Scholar] [CrossRef]
- Li, S.; Zhao, S.; Hu, C.; Mao, C.; Guo, L.; Yu, H.; Yu, H. Whole Genome Sequence of an Edible Mushroom Stropharia rugosoannulata (Daqiugaigu). J. Fungi 2022, 8, 99. [Google Scholar] [CrossRef]
- Hess, J.; Skrede, I.; Wolfe, B.E.; Labutti, K.; Ohm, R.A.; Grigoriev, I.V.; Pringle, A. Transposable element dynamics among asymbiotic and ectomycorrhizal Amanita fungi. Genome Biol. Evol. 2014, 6, 1564–1578. [Google Scholar] [CrossRef]
- Gupta, D.K.; Rühl, M.; Mishra, B.; Kleofas, V.; Hofrichter, M.; Herzog, R.; Pecyna, M.J.; Sharma, R.; Kellner, H.; Hennicke, F.; et al. The genome sequence of the commercially cultivated mushroom Agrocybe aegerita reveals a conserved repertoire of fruiting-related genes and a versatile suite of biopolymer-degrading enzymes. BMC Genom. 2018, 19, 48. [Google Scholar] [CrossRef]
- Liang, Y.; Lu, D.; Wang, S.; Zhao, Y.; Gao, S.; Han, R.; Yu, J.; Zheng, W.; Geng, J.; Hu, S. Genome Assembly and Pathway Analysis of Edible Mushroom Agrocybe cylindracea. Genom. Proteom. Bioinform. 2020, 18, 341–351. [Google Scholar] [CrossRef]
- Duan, Y.; Han, H.; Qi, J.; Gao, J.; Xu, Z.; Wang, P.; Zhang, J.; Liu, C. Genome sequencing of Inonotus obliquus reveals insights into candidate genes involved in secondary metabolite biosynthesis. BMC Genom. 2022, 23, 314. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J.; Kronstad, J.W.; Taylor, J.W.; Casselton, L.A. Sex in Fungi: Molecular Determination and Evolutionary Implications; American Association for Microbiology Press: Washington, DC, USA, 2007; ISBN 1555814212. [Google Scholar]
- Wang, G.; Wang, Y.; Chen, L.; Wang, H.; Guo, L.; Zhou, X.; Dou, M.; Wang, B.; Lin, J.; Liu, L.; et al. Genetic structure and evolutionary diversity of mating-type (MAT) loci in Hypsizygus marmoreus. IMA Fungus 2021, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Kües, U.; Casselton, L.A. Homeodomains and regulation of sexual development in basidiomycetes. Trends Genet. 1992, 8, 154. [Google Scholar] [CrossRef]
- Bao, D. Research progress on the mating-typing locus structures of basidiomycete mushrooms. Mycosystema 2019, 38, 2061–2077. [Google Scholar] [CrossRef]
- Shang, J.; Hou, D.; Li, Y.; Zhou, C.; Guo, T.; Tang, L.; Mao, W.; Chen, Q.; Bao, D.; Yang, R. Analyses of mating systems in Stropharia rugosoannulata based on genomic data. Mycosystema 2020, 39, 1152–1161. [Google Scholar] [CrossRef]
- Chen, W.; Chai, H.; Yang, W.; Ma, Y.; Zhao, Y. Characterization of B Mating Loci Identification and Application in Cyclocybe aegerita and C. salicacola. Biotechnol. Bull. 2021, 37, 57–64. [Google Scholar] [CrossRef]
- Chen, W.; Chai, H.; Yang, W.; Zhang, X.; Chen, Y.; Zhao, Y. Characterization of Non-coding Regions in B Mating Loci of Agrocybe salicacola Groups: Target Sites for B Mating Type Identification. Curr. Microbiol. 2017, 74, 772–778. [Google Scholar] [CrossRef]
- Yang, W.; Zhao, Y.; Chen, Y.; Chen, W. Cloning Analysis of Mitochondrial Intermediate Peptidase Encoding Gene and Its Application on A Mating-locus Identification in Agrocybe aegerita. North. Hortic. 2018, 12, 150–156. [Google Scholar]
- Zhu, L.; Qian, R.; Zhao, Y.; Chen, W.; Chen, Y. The Singleness Analysis of Monokaryon from Agrocybe aegerita. Chin. Agric. Sci. Bull. 2019, 35, 68–72. [Google Scholar]
- Qian, R.; Chen, W.; Chai, H.; Tao, N.; Zhao, Y.; Chen, Y. B Mating Locus from Agrocybe aegerita: Structural Analysis. Chin. Agric. Sci. Bull. 2021, 37, 14–19. [Google Scholar]
- Chen, X.; Wei, Y.; Meng, G.; Wang, M.; Peng, X.; Dai, J.; Dong, C.; Huo, G. Telomere-to-Telomere Haplotype-Resolved Genomes of Agrocybe chaxingu Reveals Unique Genetic Features and Developmental Insights. J. Fungi 2024, 10, 602. [Google Scholar] [CrossRef] [PubMed]
- May, T.W. Procedures and timetable for proposals to amend Chapter F of the International Code of Nomenclature for algae, fungi, and plants. IMA Fungus 2020, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, L.; Shang, X.; Song, C.; Yu, H.; Zhang, M.; Tan, Q. A New Method for Protoplast Preparation in Edible Fungi. Acta Edulis Fungi 2020, 27, 108–114. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Sun, P.; Hu, J.; Chen, X.; Wei, Y. Protoplast regeneration and monokaryotation characteristics of Cyclocybe aegerita. Microbiology+ 2022, 49, 2076–2087. [Google Scholar] [CrossRef]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Hu, J.; Wang, Z.; Sun, Z.; Hu, B.; Ayoola, A.O.; Liang, F.; Li, J.; Sandoval, J.R.; Cooper, D.N.; Ye, K.; et al. NextDenovo: An efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 2024, 25, 107. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simao, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Scalzitti, N.; Jeannin-Girardon, A.; Collet, P.; Poch, O.; Thompson, J.D. A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. BMC Genom. 2020, 21, 293. [Google Scholar] [CrossRef]
- Baker, L.; David, C.; Jacobs, D.J.; Mulder, N. Ab initio gene prediction for protein-coding regions. Bioinform. Adv. 2023, 3, vbad105. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids. Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids. Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids. Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids. Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids. Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernandez-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids. Res. 2018, 46, 2699. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids. Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids. Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids. Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids. Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids. Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Frings, R.A.; Macia-Vicente, J.G.; Busse, S.; Cmokova, A.; Kellner, H.; Hofrichter, M.; Hennicke, F. Multilocus phylogeny- and fruiting feature-assisted delimitation of European Cyclocybe aegerita from a new Asian species complex and related species. Mycol. Prog. 2020, 19, 1001–1016. [Google Scholar] [CrossRef]
- Teh, B.T.; Lim, K.; Yong, C.H.; Ng, C.C.Y.; Rao, S.R.; Rajasegaran, V.; Lim, W.K.; Ong, C.K.; Chan, K.; Cheng, V.K.Y.; et al. The draft genome of tropical fruit durian (Durio zibethinus). Nat. Genet. 2017, 49, 1633–1641. [Google Scholar] [CrossRef]
- Kim, M.; Oh, H.; Park, S.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef]
- Garron, M.; Henrissat, B. The continuing expansion of CAZymes and their families. Curr. Opin. Chem. Biol. 2019, 53, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Pallister, E.; Gray, C.J.; Flitsch, S.L. Enzyme promiscuity of carbohydrate active enzymes and their applications in biocatalysis. Curr. Opin. Struct. Biol. 2020, 65, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martinez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef]
- Manavalan, T.; Manavalan, A.; Heese, K. Characterization of lignocellulolytic enzymes from white-rot fungi. Curr. Microbiol. 2015, 70, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, N.; Sugiyama, M.; Igarashi, K. Random forest machine-learning algorithm classifies white-and brown-rot fungi according to the number of the genes encoding Carbohydrate-Active enZyme families. Appl. Environ. Microbiol. 2024, 90, e00424–e00482. [Google Scholar] [CrossRef]
- Suryadi, H.; Judono, J.J.; Putri, M.R.; Eclessia, A.D.; Ulhaq, J.M.; Agustina, D.N.; Sumiati, T. Biodelignification of lignocellulose using ligninolytic enzymes from white-rot fungi. Heliyon 2022, 8, e08865. [Google Scholar] [CrossRef]
- Ding, S.; Ge, W.; Buswell, J.A. Molecular cloning and transcriptional expression analysis of an intracellular beta-glucosidase, a family 3 glycosyl hydrolase, from the edible straw mushroom, Volvariella volvacea. FEMS Microbiol. Lett. 2007, 267, 221–229. [Google Scholar] [CrossRef]
- Tao, Y.; Xie, B.; Yang, Z.; Chen, Z.; Chen, B.; Deng, Y.; Jiang, Y.; van Peer, A.F. Identification and expression analysis of a new glycoside hydrolase family 55 exo-beta-1,3-glucanase-encoding gene in Volvariella volvacea suggests a role in fruiting body development. Gene 2013, 527, 154–160. [Google Scholar] [CrossRef]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef]
- Bi, Y.; Hubbard, C.; Purushotham, P.; Zimmer, J. Insights into the structure and function of membrane-integrated processive glycosyltransferases. Curr. Opin. Struct. Biol. 2015, 34, 78–86. [Google Scholar] [CrossRef]
- Greenfield, J.J.; High, S. The Sec61 complex is located in both the ER and the ER-Golgi intermediate compartment. J. Cell Sci. 1999, 112 Pt 10, 1477–1486. [Google Scholar] [CrossRef]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids. Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef] [PubMed]
- Ragucci, S.; Landi, N.; Russo, R.; Valletta, M.; Pedone, P.V.; Chambery, A.; Di Maro, A. Ageritin from Pioppino Mushroom: The Prototype of Ribotoxin-Like Proteins, a Novel Family of Specific Ribonucleases in Edible Mushrooms. Toxins 2021, 13, 263. [Google Scholar] [CrossRef] [PubMed]
- Landi, N.; Ragucci, S.; Russo, R.; Valletta, M.; Pizzo, E.; Ferreras, J.M.; Di Maro, A. The ribotoxin-like protein Ostreatin from Pleurotus ostreatus fruiting bodies: Confirmation of a novel ribonuclease family expressed in basidiomycetes. Int. J. Biol. Macromol. 2020, 161, 1329–1336. [Google Scholar] [CrossRef]
- Landi, N.; Ragucci, S.; Culurciello, R.; Russo, R.; Valletta, M.; Pedone, P.V.; Pizzo, E.; Di Maro, A. Ribotoxin-like proteins from Boletus edulis: Structural properties, cytotoxicity and in vitro digestibility. Food Chem. 2021, 359, 129931. [Google Scholar] [CrossRef] [PubMed]
- van Wetter, M.A.; Schuren, F.H.; Schuurs, T.A.; Wessels, J.G. Targeted mutation of the SC3 hydrophobin gene of Schizophyllum commune affects formation of aerial hyphae. FEMS Microbiol. Lett. 1996, 140, 265–269. [Google Scholar] [CrossRef]
- Duan, M.; Long, S.; Wu, X.; Feng, B.; Qin, S.; Li, Y.; Li, X.; Li, C.; Zhao, C.; Wang, L. Genome, transcriptome, and metabolome analyses provide new insights into the resource development in an edible fungus Dictyophora indusiata. Front. Microbiol. 2023, 14, 1137159. [Google Scholar] [CrossRef]
- Liu, C.; Bi, J.; Kang, L.; Zhou, J.; Liu, X.; Liu, Z.; Yuan, S. The molecular mechanism of stipe cell wall extension for mushroom stipe elongation growth. Fungal Biol. Rev. 2021, 35, 14–26. [Google Scholar] [CrossRef]
- Gartner, A.; Engebrecht, J. DNA repair, recombination, and damage signaling. Genetics 2022, 220, iyab178. [Google Scholar] [CrossRef]
- Ozcengiz, G.; Demain, A.L. Recent advances in the biosynthesis of penicillins, cephalosporins and clavams and its regulation. Biotechnol. Adv. 2013, 31, 287–311. [Google Scholar]
- Dörter, I.; Momany, M. Fungal cell cycle: A unicellular versus multicellular comparison. Microbiol. Spectr. 2016, 4, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Savelkoul, E.J. Molecular Evolution of Meiosis Genes in Fungi. Ph.D. Thesis, The University of Iowa, Iowa City, IA, USA, 2013. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Song, H.; Su, G.; Wang, X.; Hu, H.; Zhai, Z.; Chen, M.; Zhou, J.; Yin, H.; Gao, Y.; et al. Genomic and Transcriptomic Analysis to Explore the Biological Characteristics of Cyclocybe chaxingu. Horticulturae 2025, 11, 409. https://doi.org/10.3390/horticulturae11040409
Yang Q, Song H, Su G, Wang X, Hu H, Zhai Z, Chen M, Zhou J, Yin H, Gao Y, et al. Genomic and Transcriptomic Analysis to Explore the Biological Characteristics of Cyclocybe chaxingu. Horticulturae. 2025; 11(4):409. https://doi.org/10.3390/horticulturae11040409
Chicago/Turabian StyleYang, Qiang, Haiyan Song, Ge Su, Xuncheng Wang, Haijing Hu, Zhijun Zhai, Minghui Chen, Jianping Zhou, Hua Yin, Yang Gao, and et al. 2025. "Genomic and Transcriptomic Analysis to Explore the Biological Characteristics of Cyclocybe chaxingu" Horticulturae 11, no. 4: 409. https://doi.org/10.3390/horticulturae11040409
APA StyleYang, Q., Song, H., Su, G., Wang, X., Hu, H., Zhai, Z., Chen, M., Zhou, J., Yin, H., Gao, Y., & Hu, D. (2025). Genomic and Transcriptomic Analysis to Explore the Biological Characteristics of Cyclocybe chaxingu. Horticulturae, 11(4), 409. https://doi.org/10.3390/horticulturae11040409