
Impact of Two Hexaploidizations on Distribution, Codon Bias, and Expression of Transcription Factors in Tomato Fruit Ripeness
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Species Selection and Identification of Transcription Factors Involved in Fruit Ripening
2.2. Chromosome Localization Analysis of Transcription Factors
2.3. Codon Usage Analysis of Transcription Factors Involved in Fruit Ripening
2.4. Inferences from Phylogenetic Trees Based on Genetic Data
2.5. Analysis of the Origins of Transcription Factors Based on Collinearity
2.6. Enrichment of TF Families and Expression Clustering Analysis
3. Results
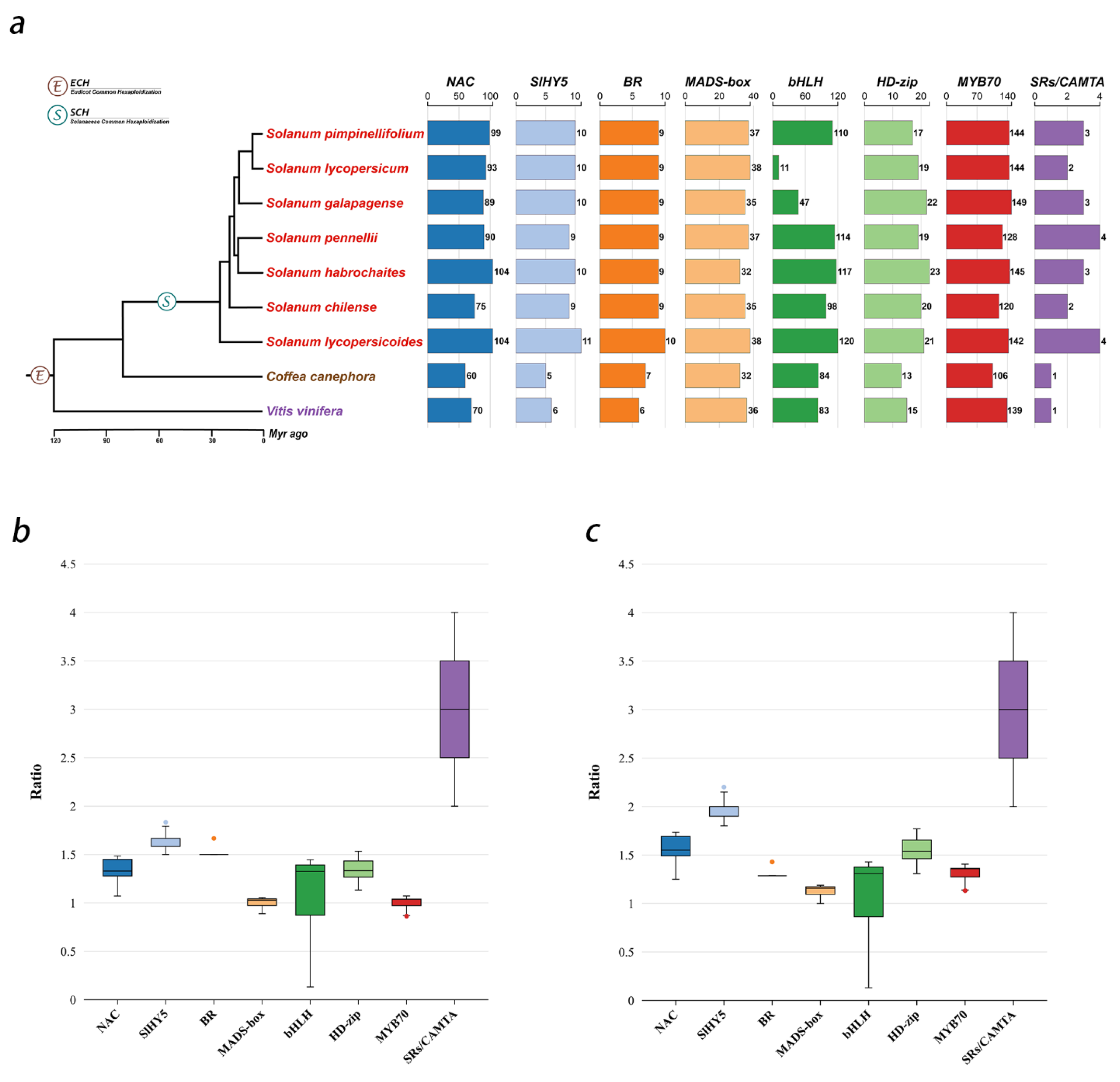
3.1. Identification of Fruit-Ripening-Related TFs and Their Quantitative Distribution Characteristics in Different Genomes
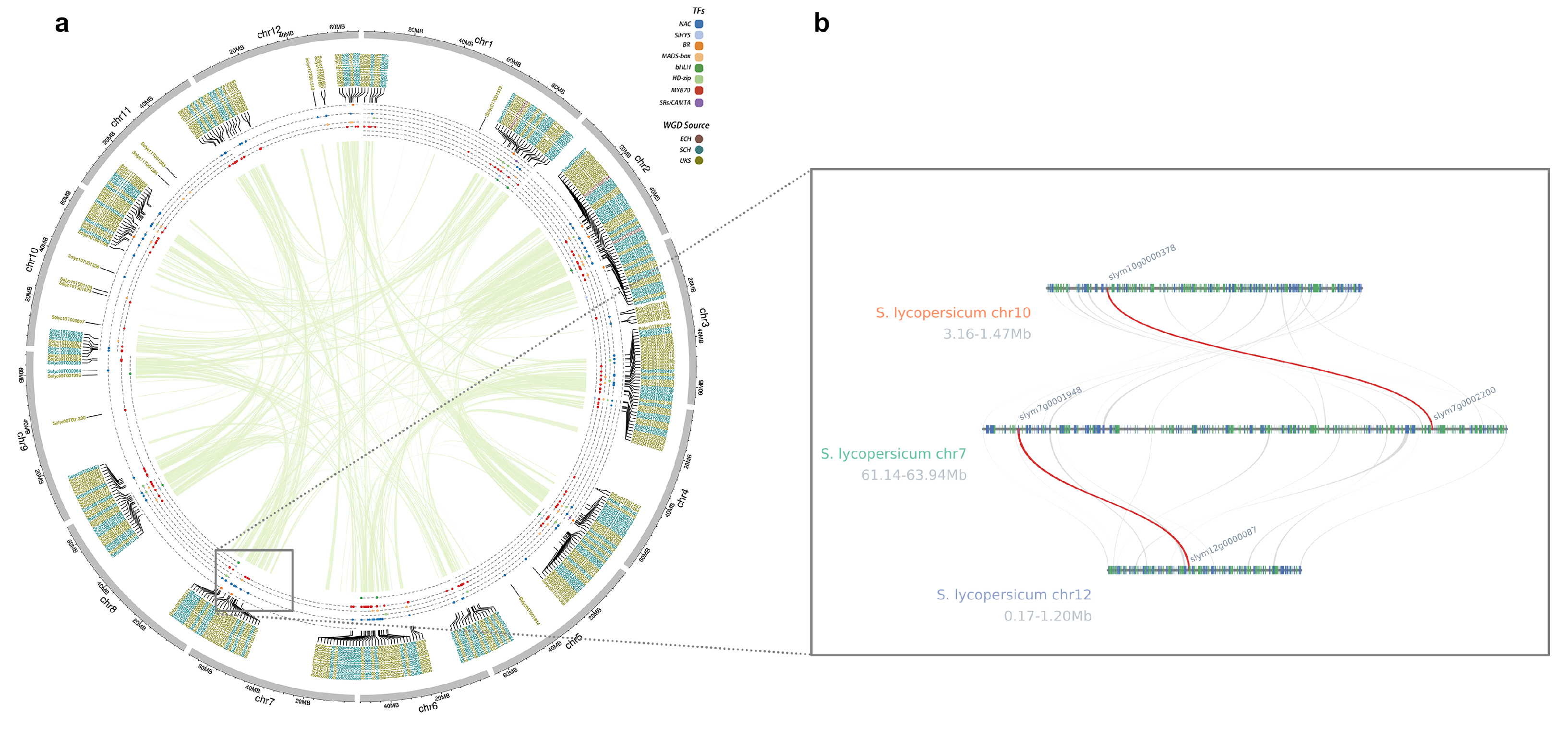
3.2. Analysis of Chromosomal Localization and Origin Characteristics of Fruit-Ripening-Related Transcription Factor Families
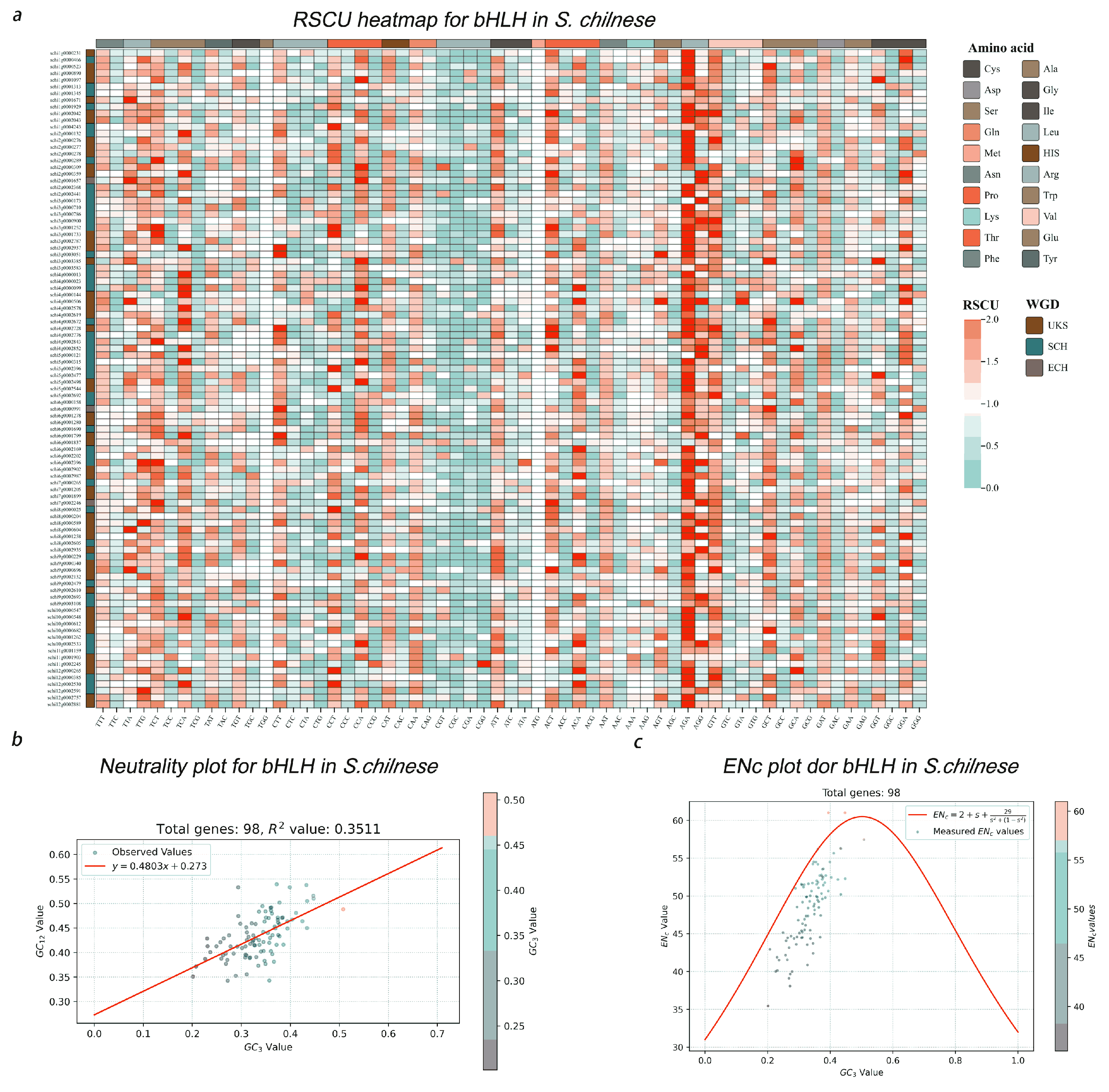
3.3. Codon Usage Bias in Fruit-Ripening-Related Transcription Factors
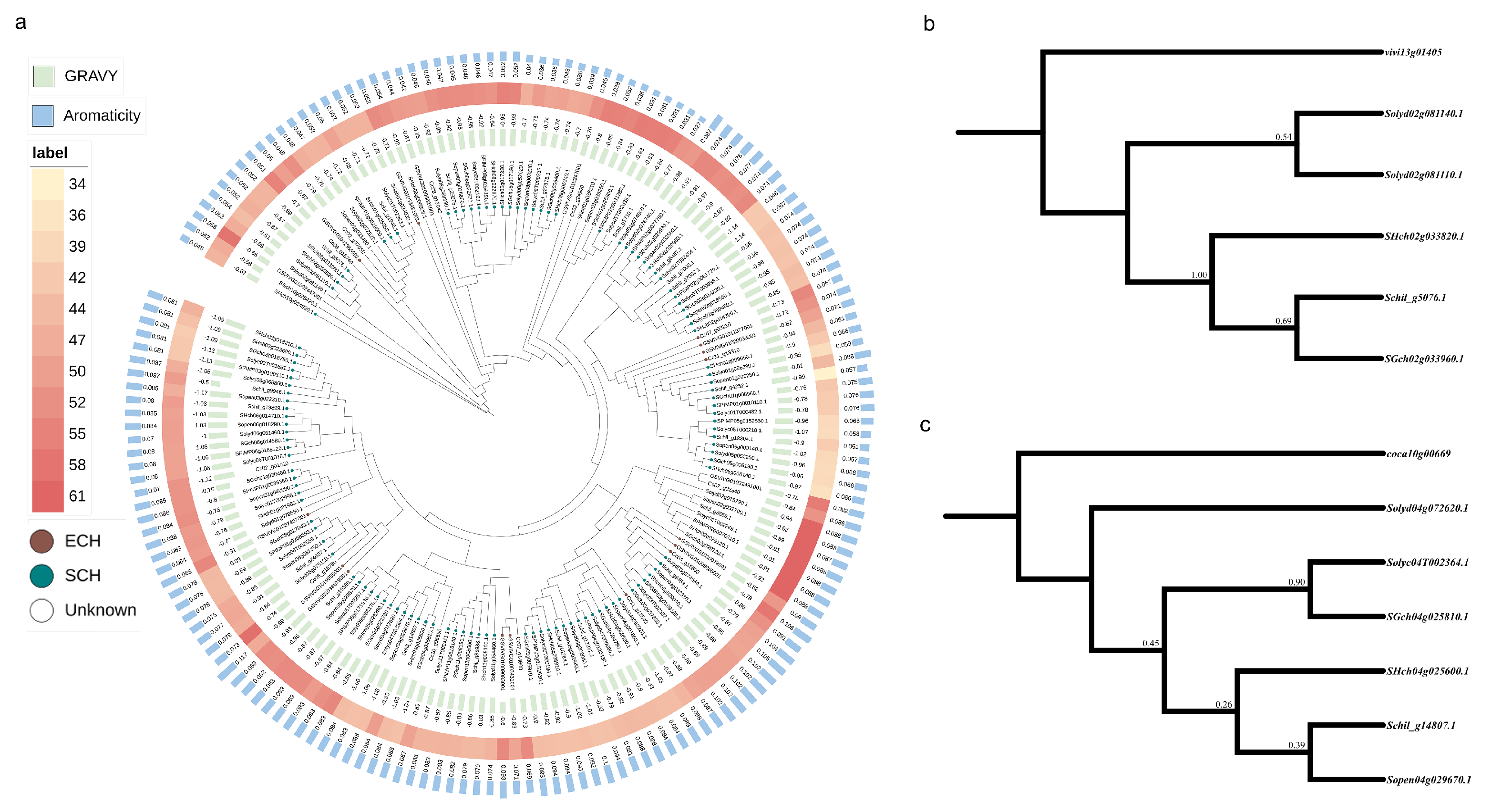
3.4. Phylogenetic Analysis of Transcription Factors in Fruit Development
3.5. Correlation Between Genomic Synteny and Fruit-Ripening-Related Transcription Factors
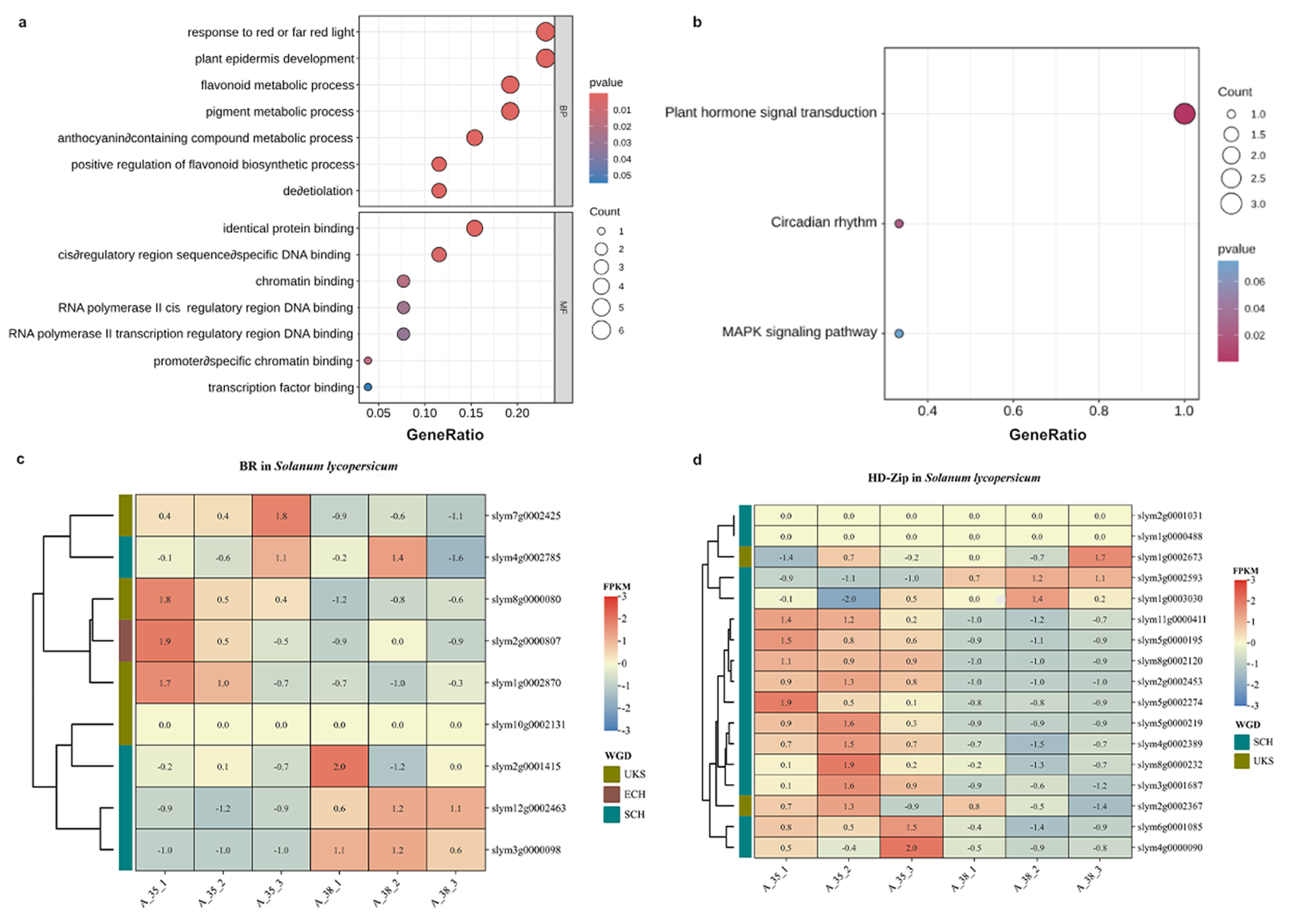
3.6. Expression Enrichment and Cluster Analysis of TF Families Related to Fruit Maturity
4. Discussion
4.1. SCH as the Primary Driver of Expansion in Specific Tomato Fruit-Ripening-Related TF Families
4.2. Polyploidization Events May Enhance Codon Usage Bias in Tomato Fruit-Ripening-Related Transcription Factor Families
4.3. Phylogenetic Analysis of Transcription Factor Family Members from Different Origins
4.4. Polyploidization May Be an Important Mechanism for Maintaining Expression Activity of Tomato Fruit-Ripening-Related Transcription Factor Families
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bergougnoux, V. The History of Tomato: From Domestication to Biopharming. Biotechnol. Adv. 2014, 32, 170–189. [Google Scholar] [CrossRef] [PubMed]
- Perveen, R.; Suleria, H.A.R.; Anjum, F.M.; Butt, M.S.; Pasha, I.; Ahmad, S. Tomato (Solanum lycopersicum) Carotenoids and Lycopenes Chemistry; Metabolism, Absorption, Nutrition, and Allied Health Claims—A Comprehensive Review. Crit. Rev. Food Sci. Nutr. 2015, 55, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Zhu, G.; Zhang, J.; Xu, X.; Yu, Q.; Zheng, Z.; Zhang, Z.; Lun, Y.; Li, S.; Wang, X.; et al. Genomic Analyses Provide Insights into the History of Tomato Breeding. Nat. Genet. 2014, 46, 1220–1226. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, C.; Ye, Z.; Li, C.; Huang, S.; Lin, T. The Genomic Route to Tomato Breeding: Past, Present, and Future. Plant Physiol. 2024, 195, 2500–2514. [Google Scholar] [CrossRef]
- Uluisik, S.; Chapman, N.H.; Smith, R.; Poole, M.; Adams, G.; Gillis, R.B.; Besong, T.M.D.; Sheldon, J.; Stiegelmeyer, S.; Perez, L.; et al. Genetic Improvement of Tomato by Targeted Control of Fruit Softening. Nat. Biotechnol. 2016, 34, 950–952. [Google Scholar] [CrossRef]
- Wang, R.; Angenent, G.C.; Seymour, G.; de Maagd, R.A. Revisiting the Role of Master Regulators in Tomato Ripening. Trends Plant Sci. 2020, 25, 291–301. [Google Scholar] [CrossRef]
- Li, C.; Hou, X.; Qi, N.; Liu, H.; Li, Y.; Huang, D.; Wang, C.; Liao, W. Insight into Ripening-Associated Transcription Factors in Tomato: A Review. Sci. Hortic. 2021, 288, 110363. [Google Scholar] [CrossRef]
- Ruprecht, C.; Lohaus, R.; Vanneste, K.; Mutwil, M.; Nikoloski, Z.; Van De Peer, Y.; Persson, S. Revisiting Ancestral Polyploidy in Plants. Sci. Adv. 2017, 3, e1603195. [Google Scholar] [CrossRef]
- Tang, H.; Bowers, J.E.; Wang, X.; Ming, R.; Alam, M.; Paterson, A.H. Synteny and Collinearity in Plant Genomes. Science 2008, 320, 486–488. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, X.; Cheng, F. Plant Polyploidy: Origin, Evolution, and Its Influence on Crop Domestication. Hortic. Plant J. 2019, 5, 231–239. [Google Scholar] [CrossRef]
- Wu, S.; Han, B.; Jiao, Y. Genetic Contribution of Paleopolyploidy to Adaptive Evolution in Angiosperms. Mol. Plant 2020, 13, 59–71. [Google Scholar] [CrossRef] [PubMed]
- The French–Italian Public Consortium for Grapevine Genome Characterization The Grapevine Genome Sequence Suggests Ancestral Hexaploidization in Major Angiosperm Phyla. Nature 2007, 449, 463–467. Available online: https://www.nature.com/articles/nature06148#citeas (accessed on 14 April 2025). [CrossRef] [PubMed]
- The Tomato Genome Consortium The Tomato Genome Sequence Provides Insights into Fleshy Fruit Evolution. Nature 2012, 485, 635–641. [CrossRef] [PubMed]
- The Potato Genome Sequencing Consortium Genome Sequence and Analysis of the Tuber Crop Potato. Nature 2011, 475, 189–195. Available online: https://www.nature.com/articles/nature10158#citeas (accessed on 14 April 2025). [CrossRef]
- Huang, J.; Xu, W.; Zhai, J.; Hu, Y.; Guo, J.; Zhang, C.; Zhao, Y.; Zhang, L.; Martine, C.; Ma, H.; et al. Nuclear Phylogeny and Insights into Whole-Genome Duplications and Reproductive Development of Solanaceae Plants. Plant Commun. 2023, 4, 100595. [Google Scholar] [CrossRef]
- Li, N.; He, Q.; Wang, J.; Wang, B.; Zhao, J.; Huang, S.; Yang, T.; Tang, Y.; Yang, S.; Aisimutuola, P.; et al. Super-Pangenome Analyses Highlight Genomic Diversity and Structural Variation across Wild and Cultivated Tomato Species. Nat. Genet. 2023, 55, 852–860. [Google Scholar] [CrossRef]
- Yu, X.; Qu, M.; Shi, Y.; Hao, C.; Guo, S.; Fei, Z.; Gao, L. Chromosome-Scale Genome Assemblies of Wild Tomato Relatives Solanum habrochaites and Solanum galapagense Reveal Structural Variants Associated with Stress Tolerance and Terpene Biosynthesis. Hortic. Res. 2022, 9, uhac139. [Google Scholar] [CrossRef]
- Powell, A.F.; Feder, A.; Li, J.; Schmidt, M.H.-W.; Courtney, L.; Alseekh, S.; Jobson, E.M.; Vogel, A.; Xu, Y.; Lyon, D.; et al. A Solanum lycopersicoides Reference Genome Facilitates Insights into Tomato Specialized Metabolism and Immunity. Plant J. 2022, 110, 1791–1810. [Google Scholar] [CrossRef]
- Bolger, A.; Scossa, F.; Bolger, M.E.; Lanz, C.; Maumus, F.; Tohge, T.; Quesneville, H.; Alseekh, S.; Sørensen, I.; Lichtenstein, G.; et al. The Genome of the Stress-Tolerant Wild Tomato Species Solanum Pennellii. Nat. Genet. 2014, 46, 1034–1038. [Google Scholar] [CrossRef]
- Wang, X.; Gao, L.; Jiao, C.; Stravoravdis, S.; Hosmani, P.S.; Saha, S.; Zhang, J.; Mainiero, S.; Strickler, S.R.; Catala, C.; et al. Genome of Solanum Pimpinellifolium Provides Insights into Structural Variants during Tomato Breeding. Nat. Commun. 2020, 11, 5817. [Google Scholar] [CrossRef]
- Denoeud, F.; Carretero-Paulet, L.; Dereeper, A.; Droc, G.; Guyot, R.; Pietrella, M.; Zheng, C.; Alberti, A.; Anthony, F.; Aprea, G.; et al. The Coffee Genome Provides Insight into the Convergent Evolution of Caffeine Biosynthesis. Science 2014, 345, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Fan, Z.; Zhang, Q.; Li, H.; Liu, G.; Jing, Y.; Zhang, Y.; Zhu, B.; Zhu, H.; Chen, J.; et al. A Tomato NAC Transcription Factor, SlNAM1, Positively Regulates Ethylene Biosynthesis and the Onset of Tomato Fruit Ripening. Plant J. 2021, 108, 1317–1331. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wei, W.; Zhao, X.; Tan, X.; Fan, Z.; Zhang, Y.; Jing, Y.; Meng, L.; Zhu, B.; Zhu, H.; et al. A NAC Transcription Factor, NOR-Like1, Is a New Positive Regulator of Tomato Fruit Ripening. Hortic. Res. 2018, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, P.; Li, X.; Wang, Y.; Tian, S.; Qin, G. The Transcription Factor SlHY5 Regulates the Ripening of Tomato Fruit at Both the Transcriptional and Translational Levels. Hortic. Res. 2021, 8, 83. [Google Scholar] [CrossRef]
- Meng, F.; Liu, H.; Hu, S.; Jia, C.; Zhang, M.; Li, S.; Li, Y.; Lin, J.; Jian, Y.; Wang, M.; et al. The Brassinosteroid Signaling Component SlBZR1 Promotes Tomato Fruit Ripening and Carotenoid Accumulation. JIPB 2023, 65, 1794–1813. [Google Scholar] [CrossRef]
- Zhu, T.; Tan, W.-R.; Deng, X.-G.; Zheng, T.; Zhang, D.-W.; Lin, H.-H. Effects of Brassinosteroids on Quality Attributes and Ethylene Synthesis in Postharvest Tomato Fruit. Postharvest Biol. Technol. 2015, 100, 196–204. [Google Scholar] [CrossRef]
- Vrebalov, J.; Ruezinsky, D.; Padmanabhan, V.; White, R.; Medrano, D.; Drake, R.; Schuch, W.; Giovannoni, J. A MADS-Box Gene Necessary for Fruit Ripening at the Tomato Ripening-Inhibitor (Rin) Locus. Science 2002, 296, 343–346. [Google Scholar] [CrossRef]
- Fujisawa, M.; Nakano, T.; Shima, Y.; Ito, Y. A Large-Scale Identification of Direct Targets of the Tomato MADS Box Transcription Factor RIPENING INHIBITOR Reveals the Regulation of Fruit Ripening. Plant Cell 2013, 25, 371–386. [Google Scholar] [CrossRef]
- Zhang, L.; Kang, J.; Xie, Q.; Gong, J.; Shen, H.; Chen, Y.; Chen, G.; Hu, Z. The Basic Helix-Loop-Helix Transcription Factor bHLH95 Affects Fruit Ripening and Multiple Metabolisms in Tomato. J. Exp. Bot. 2020, 71, 6311–6327. [Google Scholar] [CrossRef]
- Lin, Z.; Hong, Y.; Yin, M.; Li, C.; Zhang, K.; Grierson, D. A Tomato HD-zip Homeobox Protein, LeHB-1, Plays an Important Role in Floral Organogenesis and Ripening. Plant J. 2008, 55, 301–310. [Google Scholar] [CrossRef]
- Li, F.; Fu, M.; Zhou, S.; Xie, Q.; Chen, G.; Chen, X.; Hu, Z. A Tomato HD-Zip I Transcription Factor, VAHOX1, Acts as a Negative Regulator of Fruit Ripening. Hortic. Res. 2023, 10, uhac236. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Chen, J.; Yue, M.; Xu, C.; Jian, W.; Liu, Y.; Song, B.; Gao, Y.; Cheng, Y.; Li, Z. Tomato Transcriptional Repressor MYB70 Directly Regulates Ethylene-dependent Fruit Ripening. Plant J. 2020, 104, 1568–1581. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Andreeva, A.; Florentino, L.C.; Chuguransky, S.R.; Grego, T.; Hobbs, E.; Pinto, B.L.; Orr, A.; Paysan-Lafosse, T.; Ponamareva, I.; et al. InterPro: The Protein Sequence Classification Resource in 2025. Nucleic Acids Res. 2025, 53, D444–D456. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle5: High-Accuracy Alignment Ensembles Enable Unbiased Assessments of Sequence Homology and Phylogeny. Nat. Commun. 2022, 13, 6968. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER Web Server: 2018 Update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef]
- Chao, J.; Li, Z.; Sun, Y.; Aluko, O.O.; Wu, X.; Wang, Q.; Liu, G. MG2C: A User-Friendly Online Tool for Drawing Genetic Maps. Mol. Hortic. 2021, 1, 16. [Google Scholar] [CrossRef]
- Choudhuri, S.; Sau, K. CodonU: A Python Package for Codon Usage Analysis. IEEE/ACM Trans. Comput. Biol. Bioinf. 2024, 21, 36–44. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Kumar, S.; Suleski, M.; Craig, J.M.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization by One Table (tvBOT): A Web Application for Visualizing, Modifying and Annotating Phylogenetic Trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Sun, P.; Jiao, B.; Yang, Y.; Shan, L.; Li, T.; Li, X.; Xi, Z.; Wang, X.; Liu, J. WGDI: A User-Friendly Toolkit for Evolutionary Analyses of Whole-Genome Duplications and Ancestral Karyotypes. Mol. Plant 2022, 15, 1841–1851. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, L.; Tian, G.; Dong, Y.; Zhang, X.; Zhou, Z.; Luo, X.; Li, Y.; Yao, W. shinyCircos-V2.0: Leveraging the Creation of Circos Plot with Enhanced Usability and Advanced Features. iMeta 2023, 2, e109. [Google Scholar] [CrossRef]
- Tang, H.; Krishnakumar, V.; Zeng, X.; Xu, Z.; Taranto, A.; Lomas, J.S.; Zhang, Y.; Huang, Y.; Wang, Y.; Yim, W.C.; et al. JCVI: A Versatile Toolkit for Comparative Genomics Analysis. iMeta 2024, 3, e211. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium; Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The Gene Ontology Knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.; Mi, H. PANTHER: Making Genome-scale Phylogenetics Accessible to All. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Matsuura, Y.; Ishiguro-Watanabe, M. KEGG: Biological Systems Database as a Model of the Real World. Nucleic Acids Res. 2025, 53, D672–D677. [Google Scholar] [CrossRef] [PubMed]
- Martin Morgan [Cre], M.C. [Ctb] AnnotationHub: Client to Access AnnotationHub Resources, R Package Version 3.16.0; 2025. Available online: https://bioconductor.org/packages/release/bioc/html/AnnotationHub.html (accessed on 14 April 2025).
- Yu, G.; Wang, L.-G.; Yan, G.-R.; He, Q.-Y. DOSE: An R/Bioconductor Package for Disease Ontology Semantic and Enrichment Analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Xu, S.; Hu, E.; Cai, Y.; Xie, Z.; Luo, X.; Zhan, L.; Tang, W.; Wang, Q.; Liu, B.; Wang, R.; et al. Using clusterProfiler to Characterize Multiomics Data. Nat. Protoc. 2024, 19, 3292–3320. [Google Scholar] [CrossRef]
- Tenenbaum, D. KEGGREST: Client-Side REST Access to the Kyoto Encyclopedia of Genes and Genomes (KEGG), R Package Version 1.48.0. 2025. Available online: https://bioconductor.org/packages/release/bioc/html/KEGGREST.html (accessed on 14 April 2025).
- Wickham, H. Data Analysis. In ggplot2; Use R! Springer International Publishing: Cham, Switzerland, 2016; pp. 189–201. [Google Scholar]
- Guangchuang, Y. Enrichplot: Visualization of Functional Enrichment Result, R Package Version 1.28.0. 2025. Available online: https://bioconductor.org/packages/release/bioc/html/enrichplot.html (accessed on 14 April 2025).
- Wang, J.; Sun, P.; Li, Y.; Liu, Y.; Yang, N.; Yu, J.; Ma, X.; Sun, S.; Xia, R.; Liu, X.; et al. An Overlooked Paleotetraploidization in Cucurbitaceae. Mol. Biol. Evol. 2018, 35, 16–26. [Google Scholar] [CrossRef]
- Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Jin, J.; Gao, G. PlantRegMap: Charting Functional Regulatory Maps in Plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, Y.; Wang, Z.; Lei, T.; Hu, Y.; Zhang, L.; Yuan, M.; Wang, J.; Li, Y. Genome-Wide Analysis of the NAC Family Associated with Two Paleohexaploidization Events in the Tomato. Life 2022, 12, 1236. [Google Scholar] [CrossRef]
- Hosmani, P.S.; Flores-Gonzalez, M.; Van De Geest, H.; Maumus, F.; Bakker, L.V.; Schijlen, E.; Van Haarst, J.; Cordewener, J.; Sanchez-Perez, G.; Peters, S.; et al. An Improved de Novo Assembly and Annotation of the Tomato Reference Genome Using Single-Molecule Sequencing, Hi-C Proximity Ligation and Optical Maps. bioRxiv 2019, 767764. [Google Scholar]
- Zhou, Y.; Zhang, Z.; Bao, Z.; Li, H.; Lyu, Y.; Zan, Y.; Wu, Y.; Cheng, L.; Fang, Y.; Wu, K.; et al. Graph Pangenome Captures Missing Heritability and Empowers Tomato Breeding. Nature 2022, 606, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Qian, Z.; Xie, L.; Wu, H.; Luo, Q.; Zhang, Q.; He, L.; Li, F. Genome-Wide Identification and Expression Pattern of MYB Family Transcription Factors in Erianthus Fulvus. Genes 2023, 14, 2128. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-Box Gene Family in Rice: Genome-Wide Identification, Organization and Expression Profiling during Reproductive Development and Stress. BMC Genom. 2007, 8, 242. [Google Scholar] [CrossRef]
- Zhang, T.; Lv, W.; Zhang, H.; Ma, L.; Li, P.; Ge, L.; Li, G. Genome-Wide Analysis of the Basic Helix-Loop-Helix (bHLH) Transcription Factor Family in Maize. BMC Plant Biol. 2018, 18, 235. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Y.; Hu, W.; Wu, X.; Li, X.; Luo, J.; Zhu, Z.; Wang, Z.; Liu, Y. Impact of Two Hexaploidizations on Distribution, Codon Bias, and Expression of Transcription Factors in Tomato Fruit Ripeness. Horticulturae 2025, 11, 447. https://doi.org/10.3390/horticulturae11050447
Han Y, Hu W, Wu X, Li X, Luo J, Zhu Z, Wang Z, Liu Y. Impact of Two Hexaploidizations on Distribution, Codon Bias, and Expression of Transcription Factors in Tomato Fruit Ripeness. Horticulturae. 2025; 11(5):447. https://doi.org/10.3390/horticulturae11050447
Chicago/Turabian StyleHan, Yating, Wanjie Hu, Xiuling Wu, Xinyu Li, Junxi Luo, Ziying Zhu, Zhenyi Wang, and Ying Liu. 2025. "Impact of Two Hexaploidizations on Distribution, Codon Bias, and Expression of Transcription Factors in Tomato Fruit Ripeness" Horticulturae 11, no. 5: 447. https://doi.org/10.3390/horticulturae11050447
APA StyleHan, Y., Hu, W., Wu, X., Li, X., Luo, J., Zhu, Z., Wang, Z., & Liu, Y. (2025). Impact of Two Hexaploidizations on Distribution, Codon Bias, and Expression of Transcription Factors in Tomato Fruit Ripeness. Horticulturae, 11(5), 447. https://doi.org/10.3390/horticulturae11050447