
Transcriptome Analyses Reveal Distinct Defense Strategies in Chili Plants under Soilborne Disease Intervention
Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Extract Solution Preparation
2.2. Plant Materials
2.3. Plant Sample Preparation and Identification at the Germination Stage
2.4. Plant Sample Preparation and Identification at the Seedling Stage
2.5. RNA Extraction and Transcriptome Sequencing
2.6. Quantification of Gene Expression
2.7. Differential Expression Analysis
2.8. Quantitative Real-Time PCR Validation
2.9. Statistical Analysis
3. Results
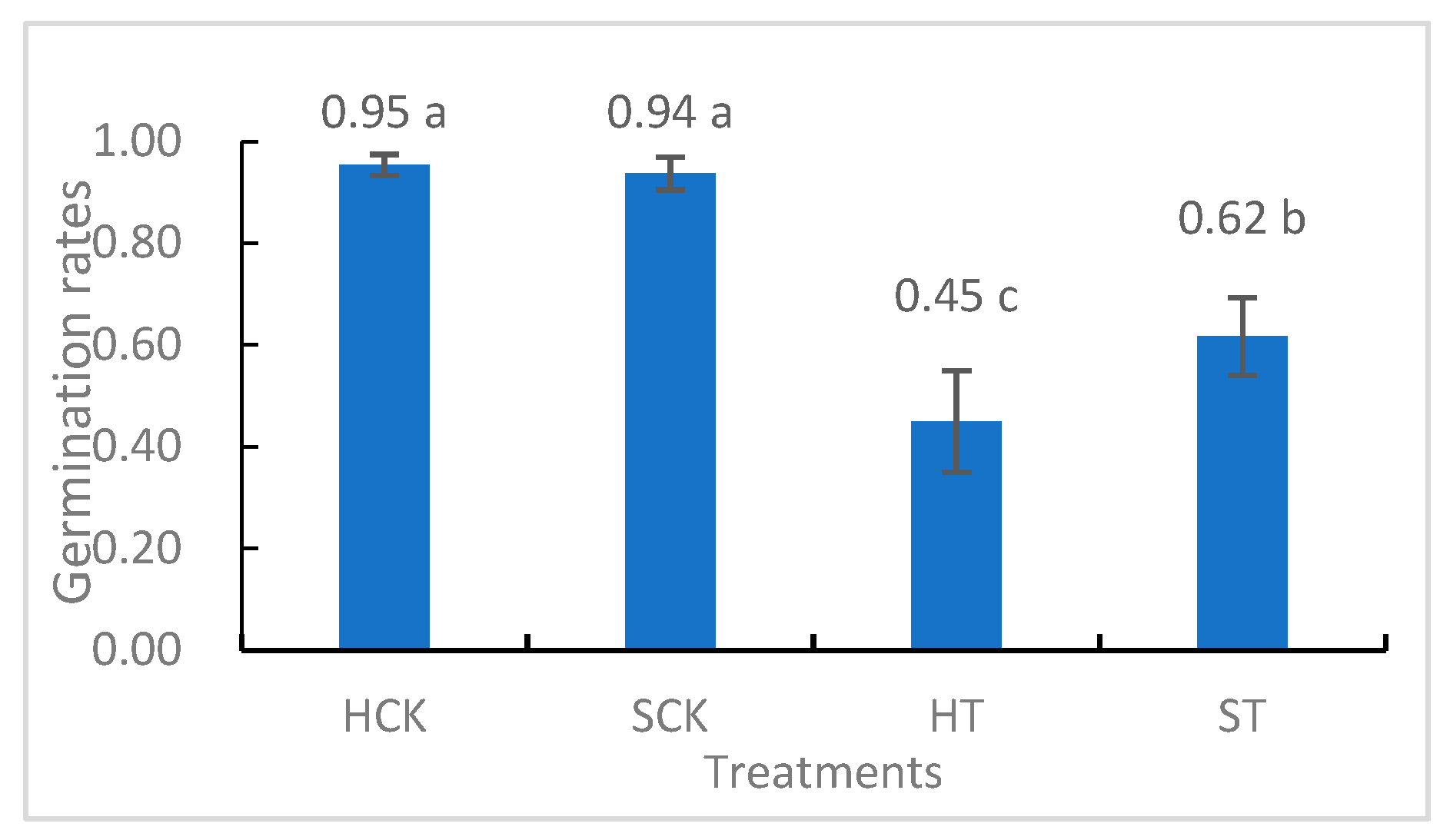
3.1. Evaluation of Germination Rates and Root Morphological Characteristics
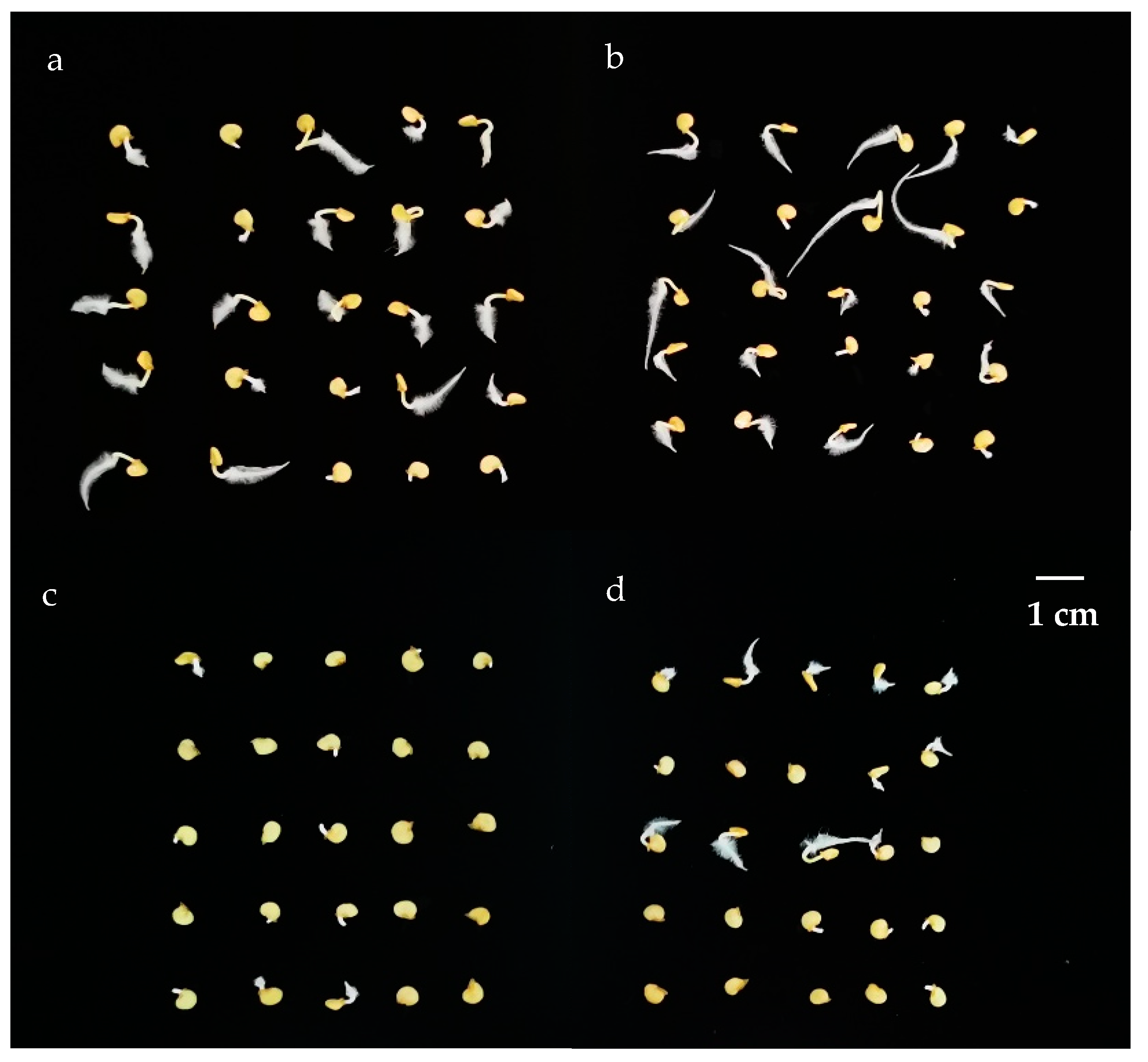
3.1.1. Germination Stage
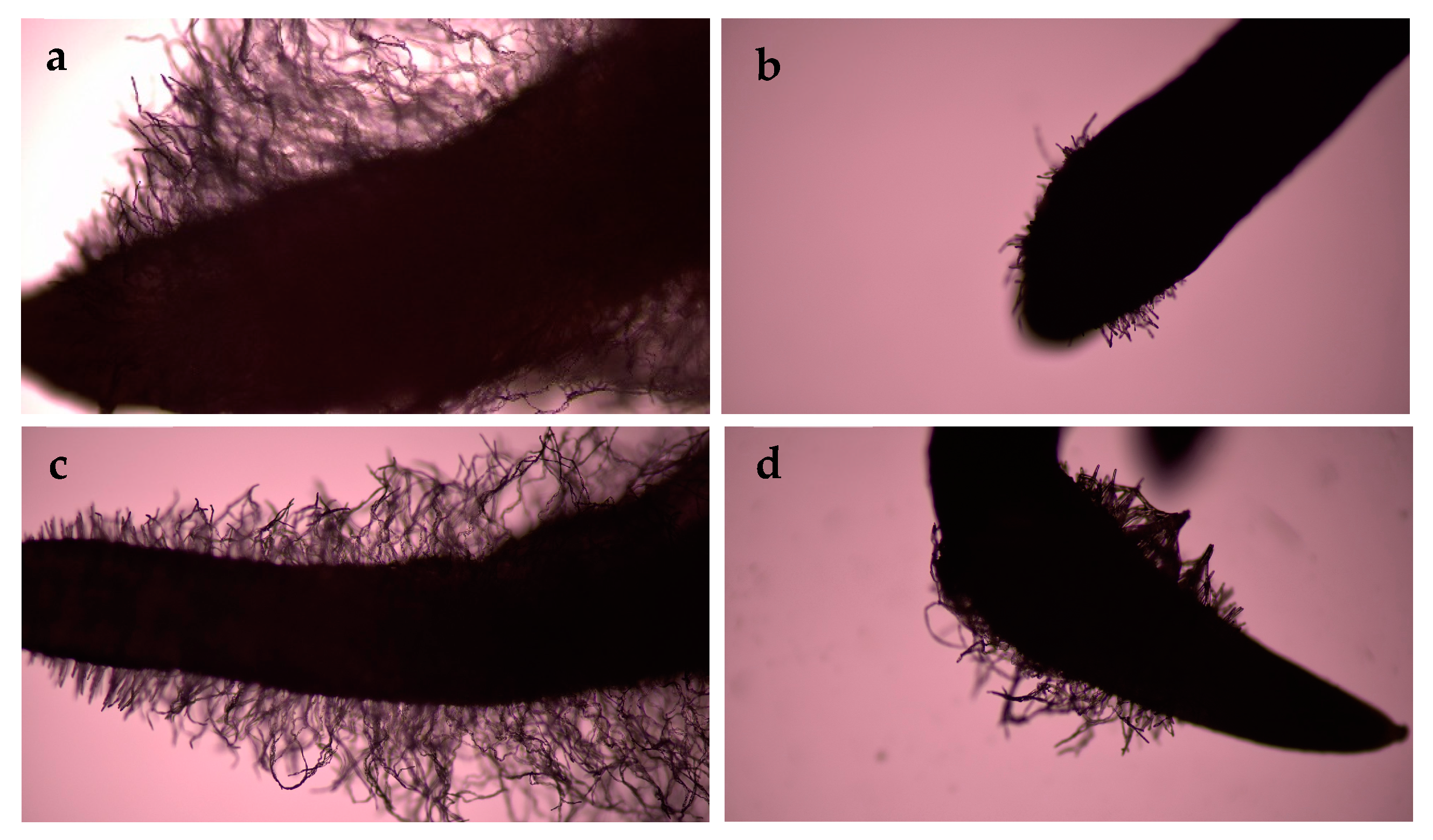
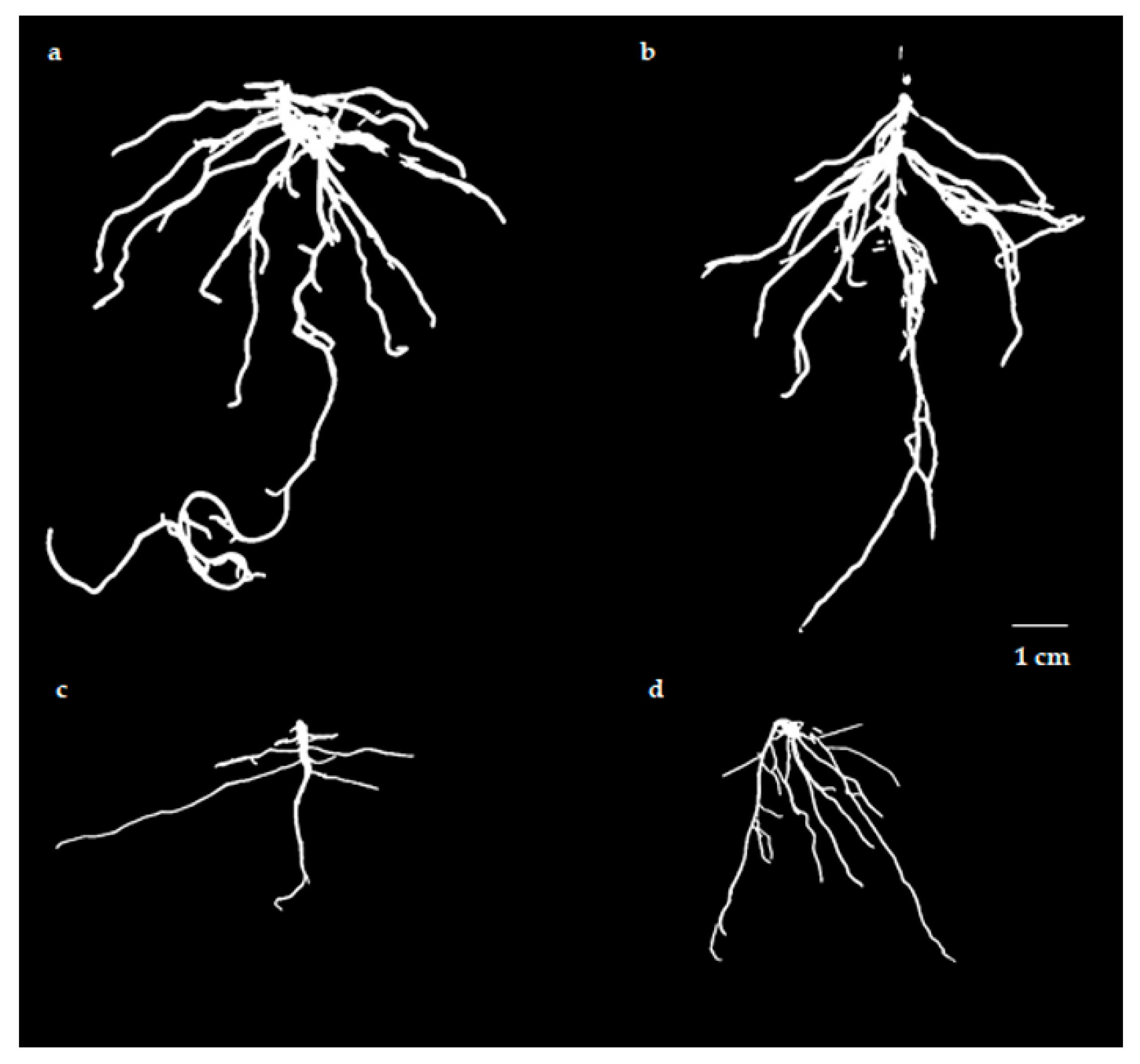
3.1.2. Seedling Stage
3.2. RNA Sequencing Analysis
3.3. Analysis of Differentially Expressed Genes (DEGs) in Chili Roots under Different Treatments
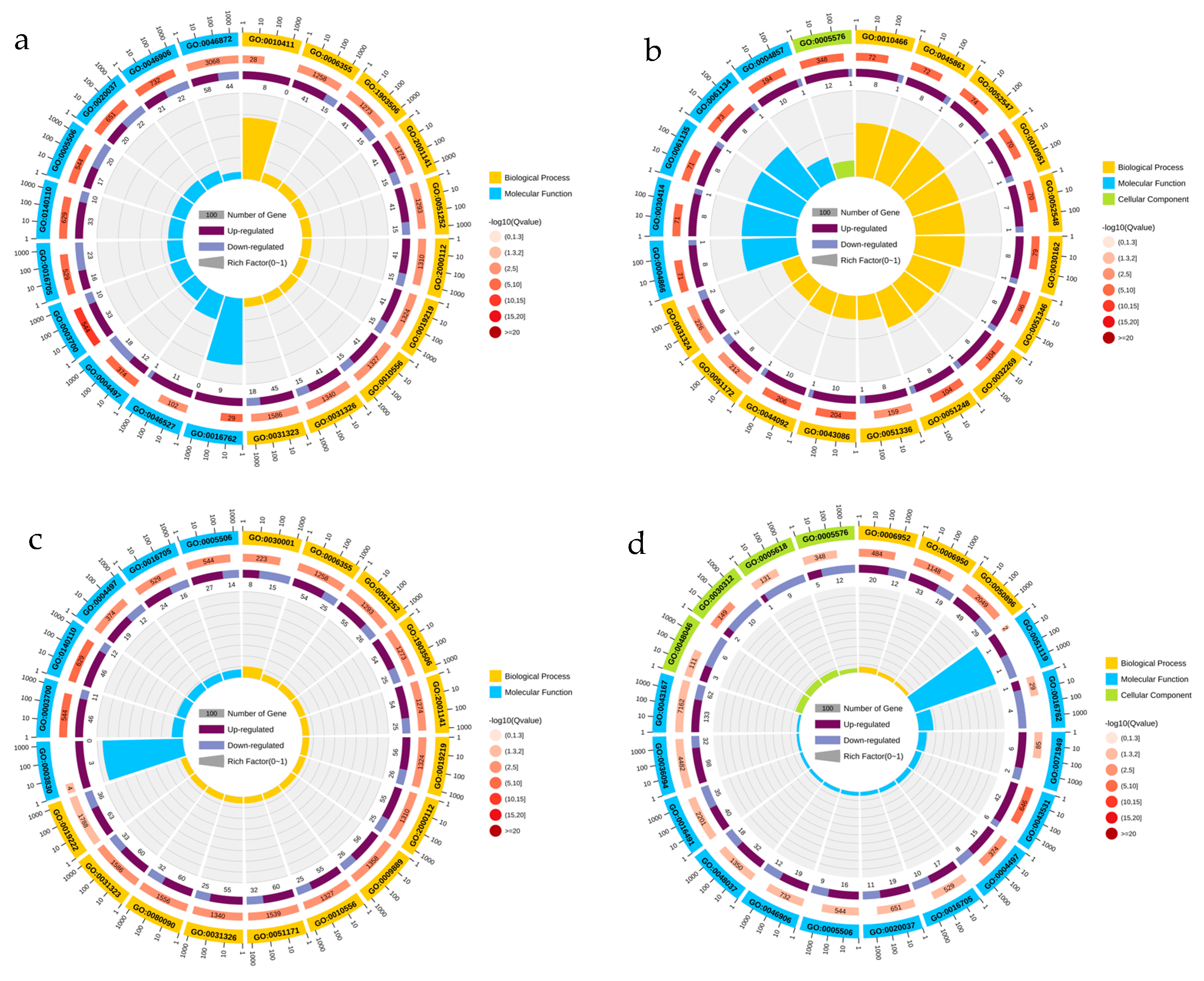
3.4. Gene Ontology Enrichment Analysis
3.5. KEGG Pathway Analysis
3.6. Screening and Analysis of DEGs in Important Biological Processes
3.6.1. DEGs Involved in Effector Recognition and Immunity Triggering in Plants
3.6.2. DEGs Related to Metabolic Processes and the Biosynthesis of Antimicrobial Secondary Metabolites
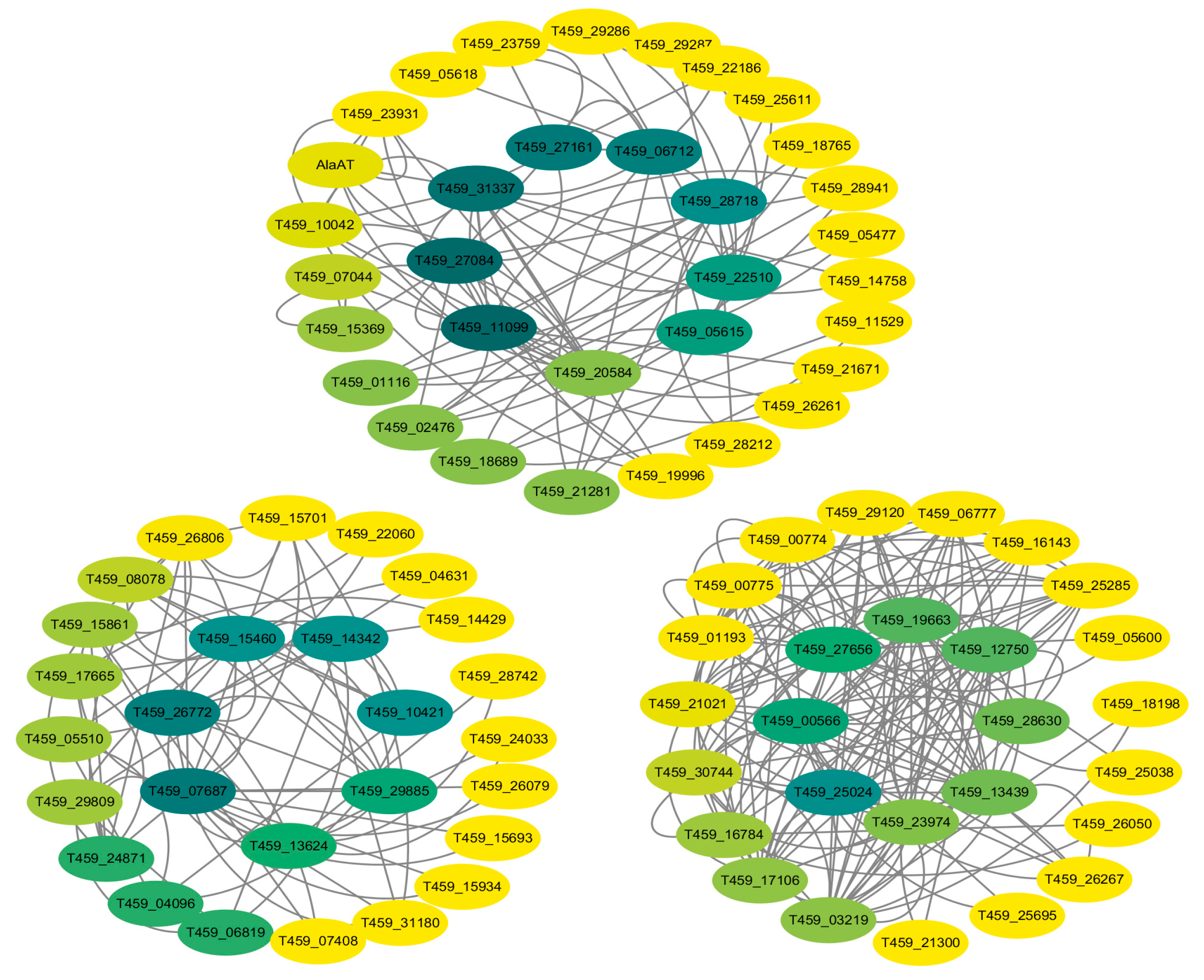
3.6.3. Co-Expression Network Analysis of the DEGs
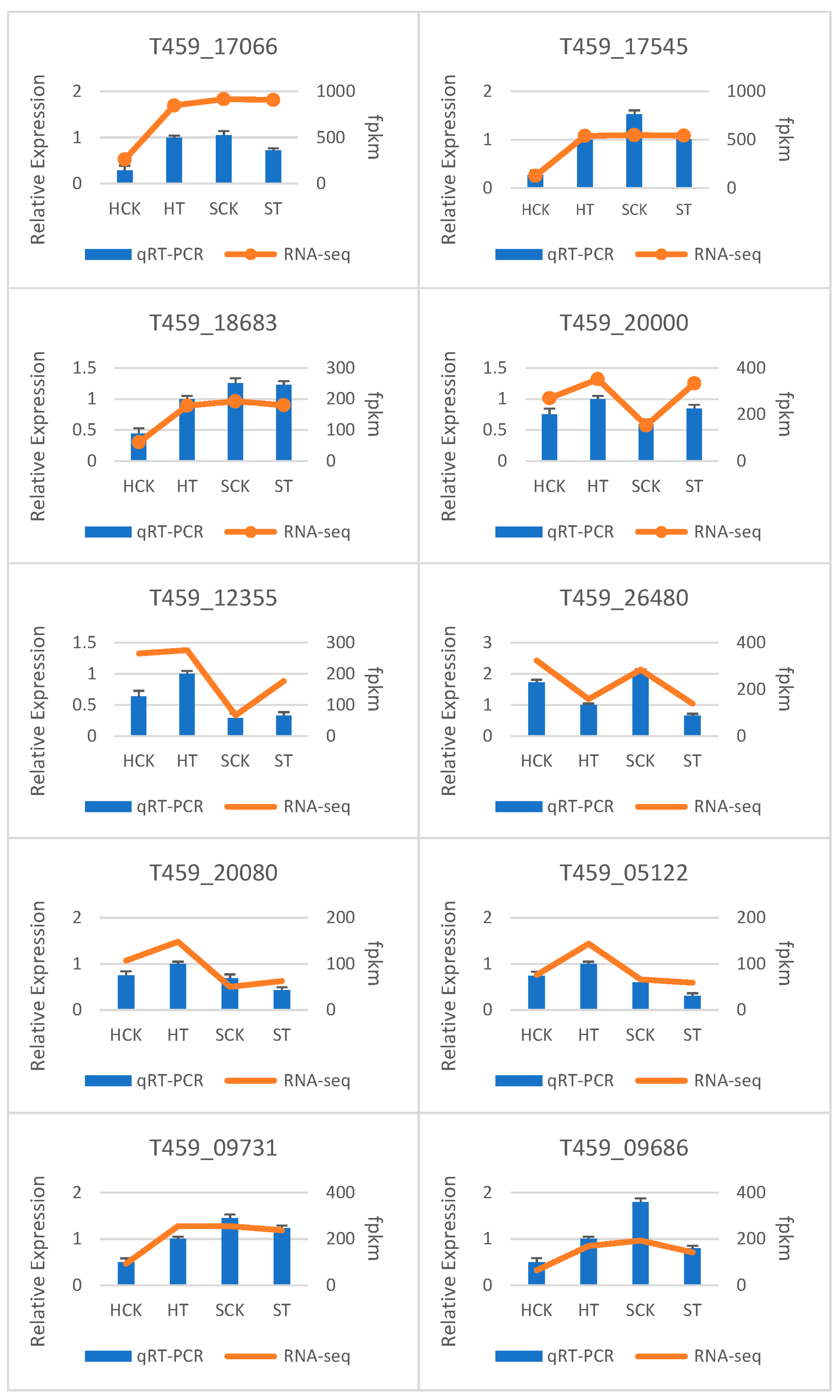
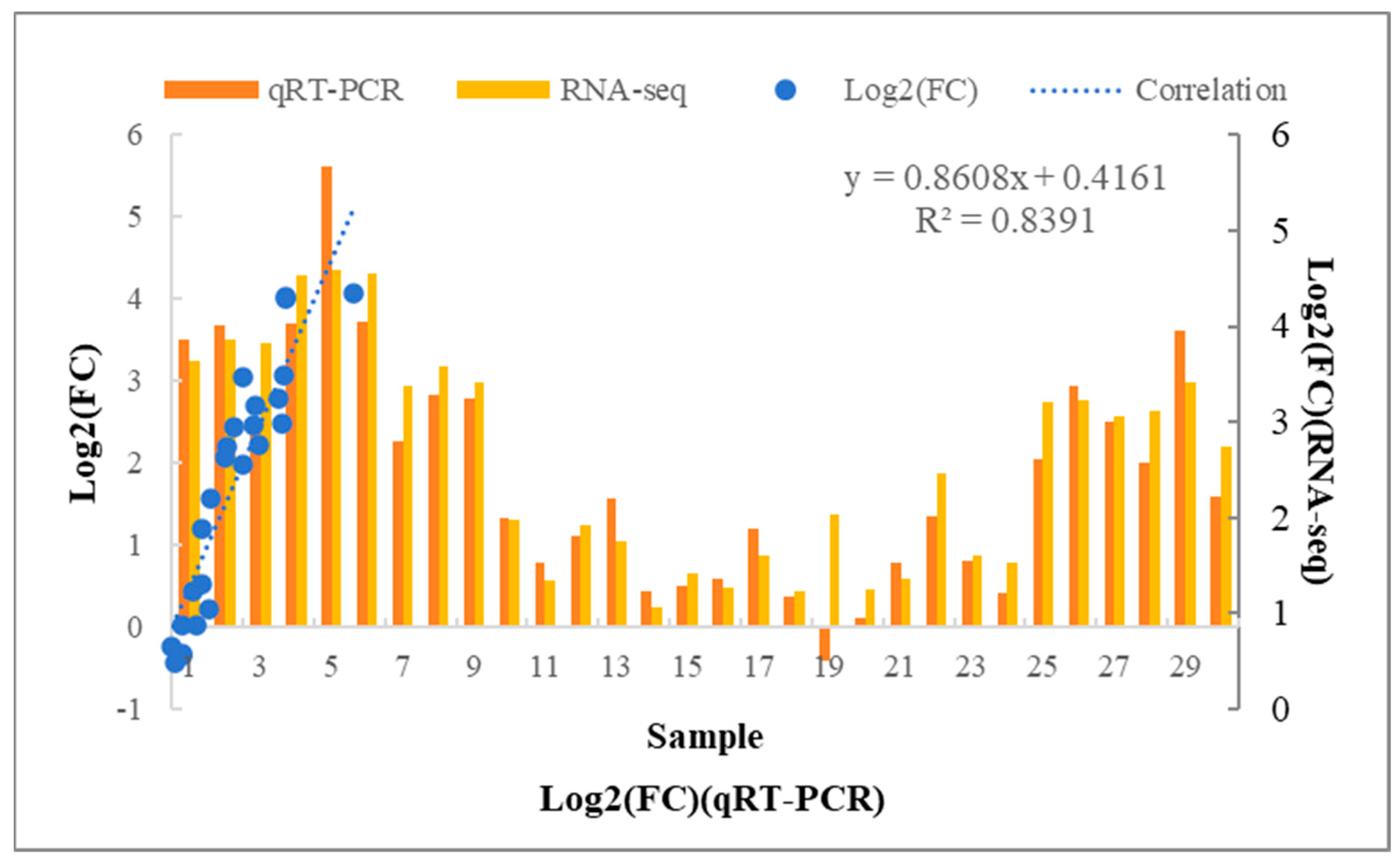
3.7. qRT-PCR Validation
4. Discussion
4.1. Root Morphological Characteristics at Different Stages
4.2. Changes in Defense Processes among Different Chili Cultivars under Stress Treatments
4.2.1. Transcriptional Changes Related to Effectors Recognized in Plants
4.2.2. Transcriptional Changes Related to Immunity Trigger in Plants
4.2.3. Transcriptional Changes Related to Modulating and Integrating the Extent of Cell Death
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jarret, R.L.; Barboza, G.E.; da Costa Batista, F.R.; Berke, T.; Chou, Y.; Hulse-Kemp, A.; Ochoa-Alejo, N.; Tripodi, P.; Veres, A.; Garcia, C.C.; et al. Capsicum-An Abbreviated Compendium. J. Am. Soc. Hortic. Sci. 2019, 144, 3–22. [Google Scholar] [CrossRef]
- Herath, H.M.S.N.; Rafii, M.Y.; Ismail, S.I.; JJ., N.; Ramlee, S.I. Improvement of Important Economic Traits in Chilli through Heterosis Breeding: A Review. J. Hortic. Sci. Biotechnol. 2021, 96, 14–23. [Google Scholar] [CrossRef]
- Karim, K.M.R.; Rafii, M.Y.; Misran, A.B.; Ismail, M.F.B.; Harun, A.R.; Khan, M.M.H.; Chowdhury, M.F.N. Current and Prospective Strategies in the Varietal Improvement of Chilli (Capsicum annuum L.) Specially Heterosis Breeding. Agronomy 2021, 11, 2217. [Google Scholar] [CrossRef]
- Yin, J.; Jackson, K.L.; Candole, B.L.; Csinos, A.S.; Langston, D.B.; Ji, P. Aggressiveness and Diversity of Phytophthora Capsici on Vegetable Crops in Georgia. Ann. Appl. Biol. 2012, 160, 191–200. [Google Scholar] [CrossRef]
- Majid, M.U.; Awan, M.F.; Fatima, K.; Tahir, M.S.; Ali, Q.; Rashid, B.; Rao, A.Q.; Nasir, I.A.; Husnain, T. Phytophthora Capsici on Chilli Pepper (Capsicum annuum L.) and Its Management through Genetic and Bio-Control: A Review. Zemdirb.-Agric. 2016, 103, 419–430. [Google Scholar] [CrossRef]
- Sanogo, S.; Ji, P. Water Management in Relation to Control of Phytophthora capsici in Vegetable Crops. Agric. Water Manag. 2013, 129, 113–119. [Google Scholar] [CrossRef]
- Naves, E.R.; de Ávila Silva, L.; Sulpice, R.; Araújo, W.L.; Nunes-Nesi, A.; Peres, L.E.P.; Zsögön, A. Capsaicinoids: Pungency beyond Capsicum. Trends Plant Sci. 2019, 24, 109–120. [Google Scholar] [CrossRef]
- de Jesus Luna-Ruiz, J.; Nabhan, G.P.; Aguilar-Meléndez, A. Shifts in Plant Chemical Defenses of Chile Pepper (Capsicum annuum L.) Due to Domestication in Mesoamerica. Front. Ecol. Evol. 2018, 6, 48. [Google Scholar] [CrossRef]
- Djian-Caporalino, C.; Palloix, A.; Fazari, A.; Marteu, N.; Barbary, A.; Abad, P.; Sage-Palloix, A.-M.; Mateille, T.; Risso, S.; Lanza, R.; et al. Pyramiding, Alternating or Mixing: Comparative Performances of Deployment Strategies of Nematode Resistance Genes to Promote Plant Resistance Efficiency and Durability. BMC Plant Biol. 2014, 14, 53. [Google Scholar] [CrossRef]
- Parisi, M.; Alioto, D.; Tripodi, P. Overview of Biotic Stresses in Pepper (Capsicum spp.): Sources of Genetic Resistance, Molecular Breeding and Genomics. Int. J. Mol. Sci. 2020, 21, 2587. [Google Scholar] [CrossRef]
- Pitrat, M. Vegetable Crops in the Mediterranean Basin with an Overview of Virus Resistance. In Advances in Virus Research, Viruses and Virus Diseases of Vegetables in the Mediterranean Basin; Génétique et Amélioration des Fruits et Légumes; Academic Press: Montfavet, France, 2012; pp. 1–29. [Google Scholar]
- Ramchiary, N.; Kehie, M.; Brahma, V.; Kumaria, S.; Tandon, P. Application of Genetics and Genomics towards Capsicum Translational Research. Plant Biotechnol. Rep. 2014, 8, 101–123. [Google Scholar] [CrossRef]
- Wang, D.; Bosland, P.W. The genes of Capsicum. HortScience 2006, 41, 1169–1187. [Google Scholar] [CrossRef]
- Feng, B.; Li, P.; Wang, H.; Zhang, X. Functional Analysis of Pcpme6 from Oomycete Plant Pathogen Phytophthora capsici. Microb. Pathog. 2010, 49, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Nonomura, T.; Appiano, M.; Pavan, S.; Matsuda, Y.; Toyoda, H.; Wolters, A.-M.A.; Visser, R.G.F.; Bai, Y. Loss of Function in Mlo Orthologs Reduces Susceptibility of Pepper and Tomato to Powdery Mildew Disease Caused by Leveillula taurica. PLoS ONE 2013, 8, e70723. [Google Scholar] [CrossRef] [PubMed]
- Sarowar, S.; Kim, E.N.; Kim, Y.J.; Ok, S.H.; Kim, K.D.; Hwang, B.K.; Shin, J.S. Overexpression of a Pepper Ascorbate Peroxidase-like 1 Gene in Tobacco Plants Enhances Tolerance to Oxidative Stress and Pathogens. Plant Sci. 2005, 169, 55–63. [Google Scholar] [CrossRef]
- Sarowar, S.; Kim, Y.J.; Kim, E.N.; Kim, K.D.; Choi, J.Y.; Hyung, N.I.; Shin, J.S. Constitutive Expression of Two Pathogenesis-Related Genes in Tomato Plants Enhanced Resistance to Oomycete Pathogen Phytophthora capsici. Plant Cell Tissue Organ Cult. 2006, 86, 7–14. [Google Scholar] [CrossRef]
- Oh, S.K.; Baek, K.H.; Seong, E.S.; Joung, Y.H.; Choi, G.J.; Park, J.M.; Cho, H.S.; Kim, E.A.; Lee, S.; Choi, D. CaMsrB2, Pepper Methionine Sulfoxide Reductase B2, Is a Novel Defense Regulator against Oxidative Stress and Pathogen Attack. Plant Physiol. 2010, 154, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Jia, Q.L.; Li, D.W.; Wang, J.E.; Yin, Y.X.; Gong, Z.H. Characteristic of the Pepper CaRGA2 Gene in Defense Responses against Phytophthora capsici Leonian. Int. J. Mol. Sci. 2013, 14, 8985–9004. [Google Scholar] [CrossRef]
- Van der Ent, S.; Van Wees, S.C.M.; Pieterse, C.M.J. Jasmonate Signaling in Plant Interactions with Resistance-Inducing Beneficial Microbes. Phytochemistry 2009, 70, 1581–1588. [Google Scholar] [CrossRef]
- Hong, J.K.; Choi, H.W.; Hwang, I.S.; Kim, D.S.; Kim, N.H.; Choi, D.S.; Kim, Y.J.; Hwang, B.K. Function of a Novel GDSL-Type Pepper Lipase Gene, CaGLIP1, in Disease Susceptibility and Abiotic Stress Tolerance. Planta 2008, 227, 539–558. [Google Scholar] [CrossRef]
- Choi, H.W.; Hwang, B.K. The Pepper Extracellular Peroxidase CaPO2 Is Required for Salt, Drought and Oxidative Stress Tolerance as Well as Resistance to Fungal Pathogens. Planta 2012, 235, 1369–1382. [Google Scholar] [CrossRef]
- Buchanan, B.; Gruissem, W.; Jones, R.L. Biochemistry & Molecular Biology of Plants; Science Press: Beijing, China, 2021; pp. 938–997, 1124–1149. [Google Scholar]
- Lee, S.C.; Hwang, B.K. CASAR82A, a Pathogen-Induced Pepper SAR8.2, Exhibits an Antifungal Activity and Its Overexpression Enhances Disease Resistance and Stress Tolerance. Plant Mol. Biol. 2006, 61, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.K.; Jeon, W.B.; Kim, K.S.; Lee, H.H.; Seo, H.H.; Kim, Y.S.; Oh, B.-J. A Colletotrichum Gloeosporioides-Induced Esterase Gene of Nonclimacteric Pepper (Capsicum annuum) Fruit during Ripening Plays a Role in Resistance against Fungal Infection. Plant Mol. Biol. 2005, 58, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.C.; Han, Y.J.; Kim, S.J.; Lee, S.S.; Kim, J.I. Resistance to rhizoctonia solani ag-2-2 (IIIB) in creeping bentgrass plants transformed with pepper esterase gene pepest. Plant Pathol. 2011, 60, 631–639. [Google Scholar] [CrossRef]
- Kraft, K.H.; Brown, C.H.; Nabhan, G.P.; Luedeling, E.; de Jesus Luna-Ruiz, J.; Coppens d’Eeckenbrugge, G.; Hijmans, R.J.; Gepts, P. Multiple Lines of Evidence for the Origin of Domesticated Chili Pepper, Capsicum annuum, in Mexico. Proc. Natl. Acad. Sci. USA 2014, 111, 6165–6170. [Google Scholar] [CrossRef] [PubMed]
- Carrizo García, C.; Barfuss, M.H.J.; Sehr, E.M.; Barboza, G.E.; Samuel, R.; Moscone, E.A.; Ehrendorfer, F. Phylogenetic Relationships, Diversification and Expansion of Chili Peppers (Capsicum, Solanaceae). Ann. Bot. 2016, 118, 35–51. [Google Scholar] [CrossRef]
- Potters, G.; Pasternak, T.P.; Guisez, Y.; Jansen, M.A.K. Different Stresses, Similar Morphogenic Responses: Integrating a Plethora of Pathways. Plant Cell Environ. 2009, 32, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Potters, G.; Pasternak, T.P.; Guisez, Y.; Palme, K.J.; Jansen, M.A.K. Stress-Induced Morphogenic Responses: Growing out of Trouble? Trends Plant Sci. 2007, 12, 98–105. [Google Scholar] [CrossRef]
- Villalobos, D.P.; Hardtke, C.S. Natural genetic variation of root system architecture from arabidopsis to brachypodium: Towards adaptive value. Philos. Trans. R. Soc. Lond. B Biol. 2012, 367, 1552–1558. [Google Scholar] [CrossRef]
- Osmont, K.S.; Sibout, R.; Hardtke, C.S. Hidden Branches: Developments in Root System Architecture. Annu. Rev. Plant Biol. 2007, 58, 93–113. [Google Scholar] [CrossRef]
- Aiken, R.M.; Smucker, A.J.M. Root System Regulation of Whole Plant Growth. Annu. Rev. Phytopathol. 1996, 34, 325–346. [Google Scholar] [CrossRef]
- Martinez, L.; Pouvreau, J.-B.; Montiel, G.; Jestin, C.; Delavault, P.; Simier, P.; Poulin, L. Soil Microbiota Promotes Early Developmental Stages of Phelipanche ramosa L. Pomel during Plant Parasitism on Brassica napus L. Plant Soil 2023, 483, 667–691. [Google Scholar] [CrossRef]
- Lechat, M.M.; Pouvreau, J.B.; Peron, T.; Gauthier, M.; Montiel, G.; Veronesi, C.; Todoroki, Y.; Le Bizec, B.; Monteau, F.; Macherel, D.; et al. PrCYP707A1, an ABA Catabolic Gene, Is a Key Component of Phelipanche Ramosa Seed Germination in Response to the Strigolactone Analogue GR24. J. Exp. Bot. 2012, 63, 5311–5322. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, M.; Yeom, S.-I.; Kim, Y.-M.; Lee, J.M.; Lee, H.-A.; Seo, E.; Choi, J.; Cheong, K.; Kim, K.-T.; et al. Genome Sequence of the Hot Pepper Provides Insights into the Evolution of Pungency in Capsicum Species. Nat. Genet. 2014, 46, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Rouhallah, S.; Choong, M.R. Revisiting bacterial volatile-mediated plant growth promotion: Lessons from the past and objectives for the future. Ann. Bot. 2018, 122, 349–358. [Google Scholar]
- Klein, S.P.; Schneider, H.M.; Perkins, A.C.; Brown, K.M.; Lynch, J.P. Multiple Integrated Root Phenotypes Are Associated with Improved Drought Tolerance. Plant Physiol. 2020, 183, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Pii, Y.; Mimmo, T.; Tomasi, N.; Terzano, R.; Cesco, S.; Crecchio, C. Microbial Interactions in the Rhizosphere: Beneficial Influences of Plant Growth-Promoting Rhizobacteria on Nutrient Acquisition Process. A Review. Biol. Fertil. Soils 2015, 51, 403–415. [Google Scholar] [CrossRef]
- Lynch, J.P. Harnessing Root Architecture to Address Global Challenges. Plant J. 2022, 109, 415–431. [Google Scholar] [CrossRef]
- Zinta, R.; Tiwari, J.K.; Buckseth, T.; Thakur, K.; Goutam, U.; Kumar, D.; Challam, C.; Bhatia, N.; Poonia, A.K.; Naik, S.; et al. Root system architecture for abiotic stress tolerance in potato: Lessons from plants. Front. Plant Sci. 2022, 13, 926214. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, X.; Li, J.; Gong, X.; Yang, P.; Gao, J.; Wang, P.; Feng, B. Comparative Analysis of Proso Millet (Panicum miliaceum L.) Leaf Transcriptomes for Insight into Drought Tolerance Mechanisms. BMC Plant Biol. 2019, 19, 397. [Google Scholar] [CrossRef]
- Belkhadir, Y.; Nimchuk, Z.; Hubert, D.A.; Mackey, D.; Dangl, J.L. Arabidopsis RIN4 Negatively Regulates Disease Resistance Mediated by RPS2 and RPM1 Downstream or Independent of the NDR1 Signal Modulator and Is Not Required for the Virulence Functions of Bacterial Type III Effectors AvrRpt2 or AvrRpm1. Plant Cell 2004, 16, 2822–2835. [Google Scholar] [CrossRef]
- Li, Z.; Huang, J.; Wang, Z.; Meng, F.; Zhang, S.; Wu, X.; Zhang, Z.; Gao, Z. Overexpression of Arabidopsis Nucleotide-Binding and Leucine-Rich Repeat Genes RPS2 and RPM1 (D505V) Confers Broad-Spectrum Disease Resistance in Rice. Front. Plant Sci. 2019, 10, 417. [Google Scholar] [CrossRef]
- Jackson, S.E. Hsp90: Structure and Function. In Topics in Current Chemistry, Molecular Chaperones; Springer: Berlin/Heidelberg, Germany, 2012; pp. 155–240. [Google Scholar]
- Khlopkov, A.; Sherstneva, O.; Ladeynova, M.; Grinberg, M.; Yudina, L.; Sukhov, V.; Vodeneev, V. Participation of Calcium Ions in Induction of Respiratory Response Caused by Variation Potential in Pea Seedlings. Plant Signal. Behav. 2021, 16, 1869415. [Google Scholar] [CrossRef] [PubMed]
- Otulak, K.; Garbaczewska, G. Cellular Localisation of Calcium Ions during Potato Hypersensitive Response to Potato Virus Y. Micron 2011, 42, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Apel, K.; Hirt, H. Reactive Oxygen Species: Metabolism, Oxidative Stress, and Signal Transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.; Schwarze, J.; Sherwood, O.L.; Jnaid, Y.; McCabe, P.F.; Kacprzyk, J. Stressed to Death: The Role of Transcription Factors in Plant Programmed Cell Death Induced by Abiotic and Biotic Stimuli. Front. Plant Sci. 2020, 11, 1235. [Google Scholar] [CrossRef]
- Bailly, A.; Groenhagen, U.; Schulz, S.; Geisler, M.; Eberl, L.; Weisskopf, L. The Inter-kingdom Volatile Signal Indole Promotes Root Development by Interfering with Auxin Signalling. Plant J. 2014, 80, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Castulo-Rubio, D.Y.; Alejandre-Ramírez, N.A.; del Carmen Orozco-Mosqueda, M.; Santoyo, G.; Macías-Rodríguez, L.I.; Valencia-Cantero, E. Volatile Organic Compounds Produced by the Rhizobacterium Arthrobacter agilis UMCV2 Modulate Sorghum bicolor (Strategy II Plant) Morphogenesis and SbFRO1 Transcription In Vitro. J. Plant Growth Regul. 2015, 34, 611–623. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatments | Root Length (cm) | Surface Area (cm2) | Average Diameter (mm) | Volume (cm3) | Total Tips | Dry Weight (g) |
|---|---|---|---|---|---|---|
| HCK | 70.7 ± 2.560 a | 24.8 ± 3.065 a | 1.17 ± 0.055 a | 0.94 ± 0.048 a | 47 ± 2.517 a | 0.015 ± 0.003 a |
| SCK | 59.8 ± 3.750 b | 22.1 ± 1.702 a | 1.16 ± 0.036 a | 0.85 ± 0.020 b | 39 ± 1.528 b | 0.014 ± 0.003 ab |
| HT | 17.2 ± 1.657 d | 5.5 ± 1.541 b | 0.62 ± 0.089 b | 0.09 ± 0.007 c | 15 ± 1.732 d | 0.009 ± 0.002 b |
| ST | 28.0 ± 1.976 c | 7.6 ± 1.630 b | 0.60 ± 0.019 b | 0.12 ± 0.013 c | 21 ± 1.528 c | 0.010 ± 0.002 ab |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Chen, Z.; Chen, F.; Yan, J.; Wu, J.; Wang, J.; Ge, S. Transcriptome Analyses Reveal Distinct Defense Strategies in Chili Plants under Soilborne Disease Intervention. Horticulturae 2023, 9, 1267. https://doi.org/10.3390/horticulturae9121267
Zhang Y, Chen Z, Chen F, Yan J, Wu J, Wang J, Ge S. Transcriptome Analyses Reveal Distinct Defense Strategies in Chili Plants under Soilborne Disease Intervention. Horticulturae. 2023; 9(12):1267. https://doi.org/10.3390/horticulturae9121267
Chicago/Turabian StyleZhang, Yuyu, Zhixiong Chen, Fang Chen, Jinqiang Yan, Junyu Wu, Jie Wang, and Shumei Ge. 2023. "Transcriptome Analyses Reveal Distinct Defense Strategies in Chili Plants under Soilborne Disease Intervention" Horticulturae 9, no. 12: 1267. https://doi.org/10.3390/horticulturae9121267